

Neuroinflammation and psychiatric illness
- PMID: 23547920
- PMCID: PMC3626880
- DOI: 10.1186/1742-2094-10-43
Neuroinflammation and psychiatric illness
Abstract
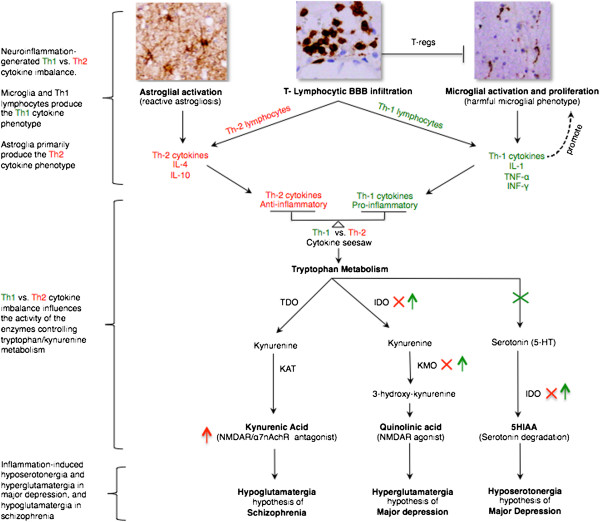
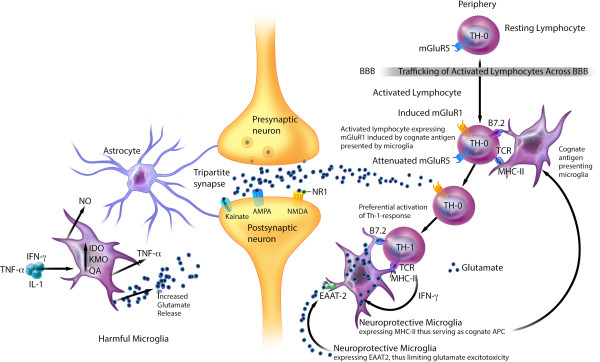
Multiple lines of evidence support the pathogenic role of neuroinflammation in psychiatric illness. While systemic autoimmune diseases are well-documented causes of neuropsychiatric disorders, synaptic autoimmune encephalitides with psychotic symptoms often go under-recognized. Parallel to the link between psychiatric symptoms and autoimmunity in autoimmune diseases, neuroimmunological abnormalities occur in classical psychiatric disorders (for example, major depressive, bipolar, schizophrenia, and obsessive-compulsive disorders). Investigations into the pathophysiology of these conditions traditionally stressed dysregulation of the glutamatergic and monoaminergic systems, but the mechanisms causing these neurotransmitter abnormalities remained elusive. We review the link between autoimmunity and neuropsychiatric disorders, and the human and experimental evidence supporting the pathogenic role of neuroinflammation in selected classical psychiatric disorders. Understanding how psychosocial, genetic, immunological and neurotransmitter systems interact can reveal pathogenic clues and help target new preventive and symptomatic therapies.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
