

Endomembrane H-Ras controls vascular endothelial growth factor-induced nitric-oxide synthase-mediated endothelial cell migration
- PMID: 23548900
- PMCID: PMC3663556
- DOI: 10.1074/jbc.M112.427765
Endomembrane H-Ras controls vascular endothelial growth factor-induced nitric-oxide synthase-mediated endothelial cell migration
Abstract
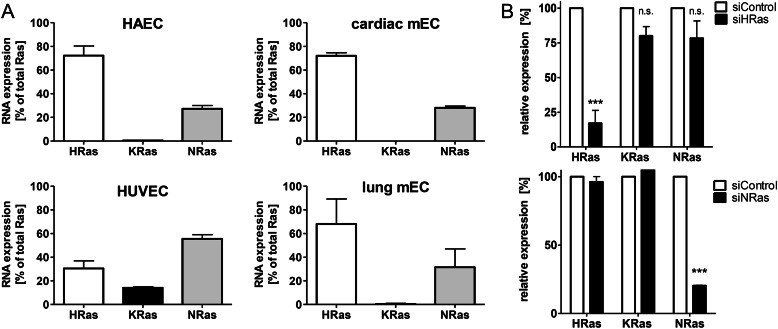
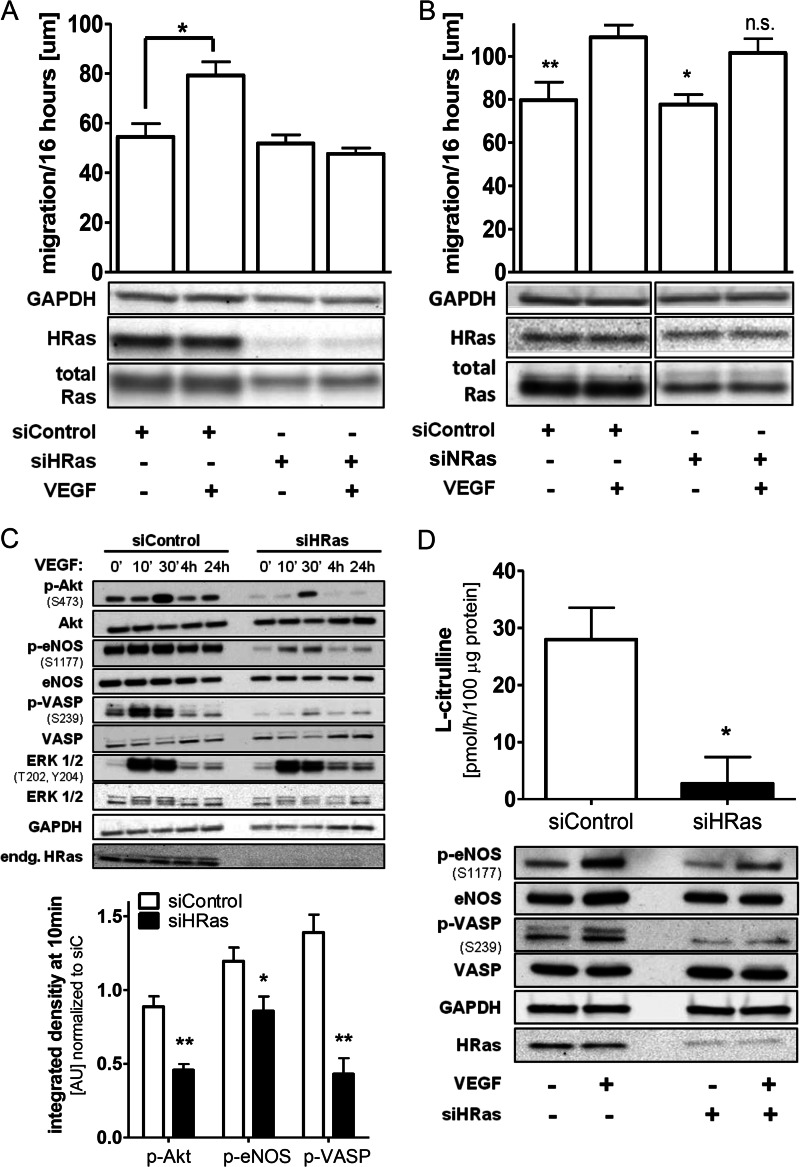
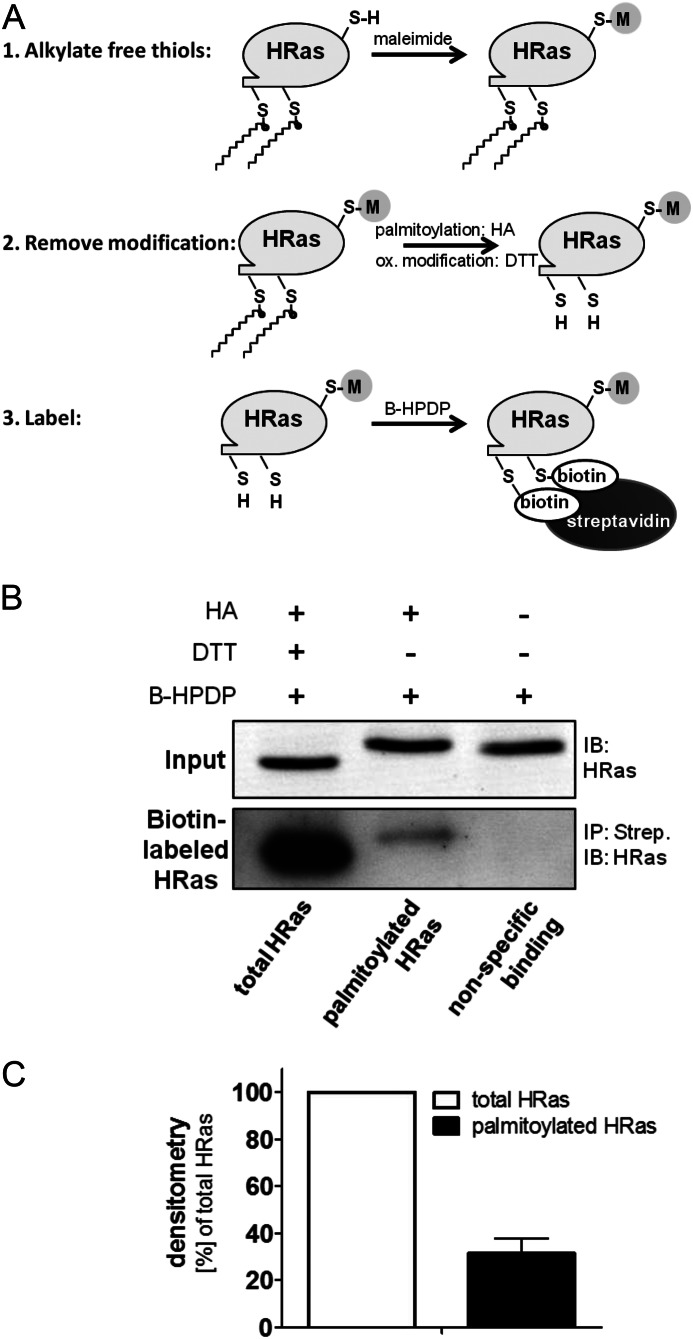
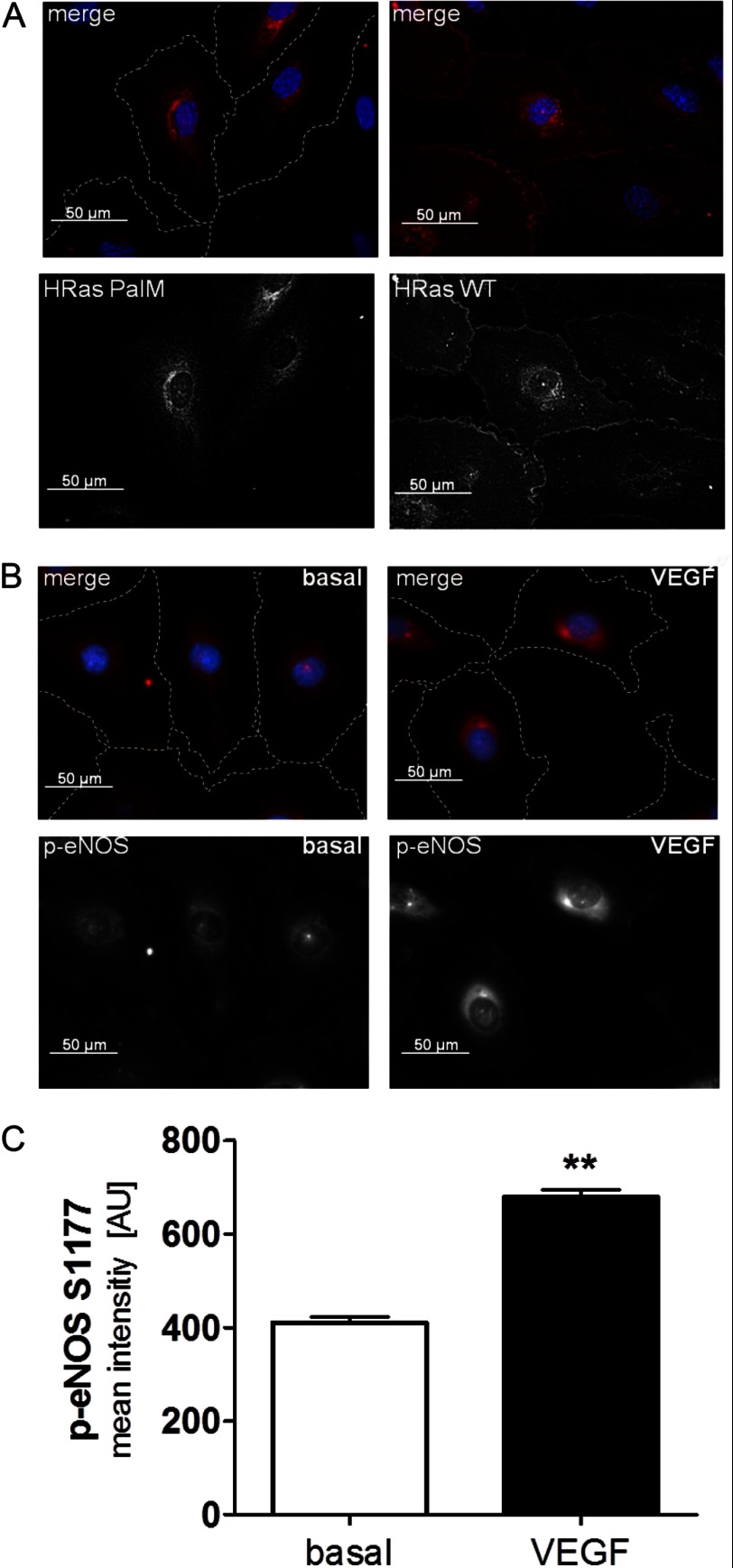
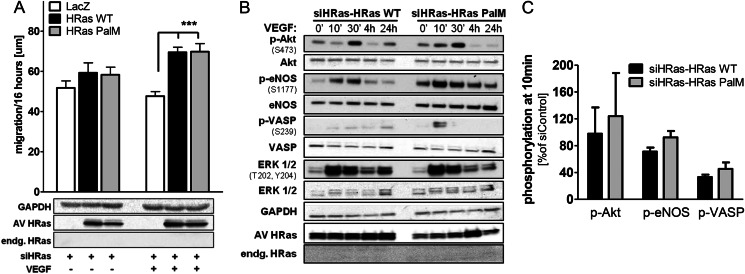
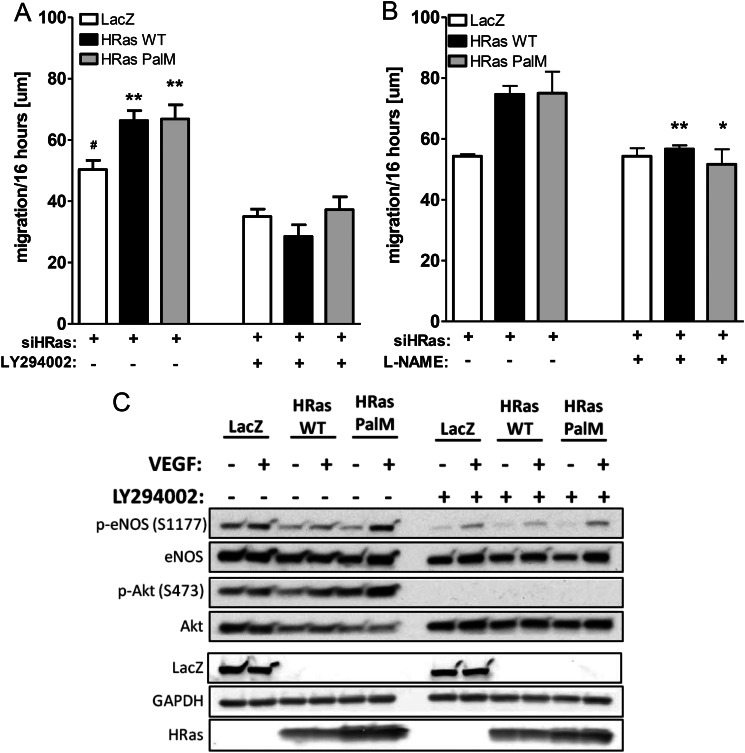
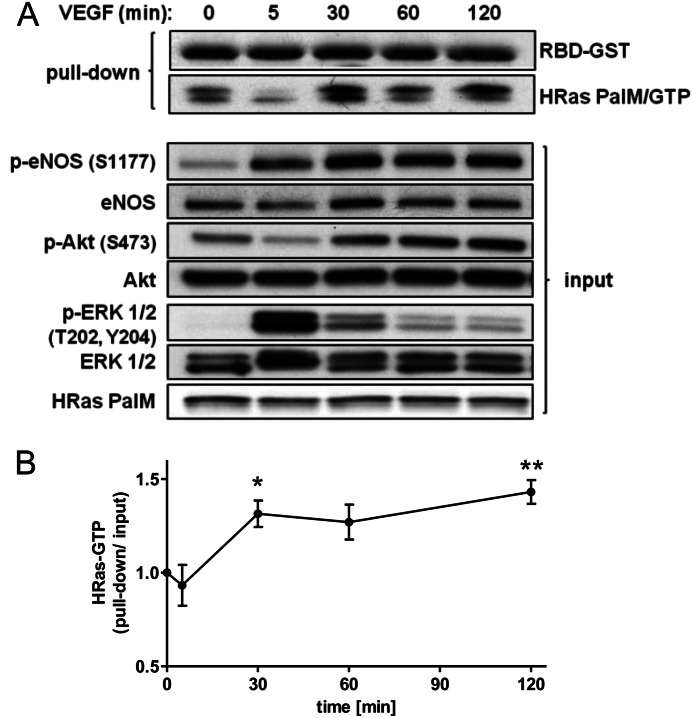
We demonstrate for the first time that endomembrane-delimited H-Ras mediates VEGF-induced activation of endothelial nitric-oxide synthase (eNOS) and migratory response of human endothelial cells. Using thiol labeling strategies and immunofluorescent cell staining, we found that only 31% of total H-Ras is S-palmitoylated, tethering the small GTPase to the plasma membrane but leaving the function of the large majority of endomembrane-localized H-Ras unexplained. Knockdown of H-Ras blocked VEGF-induced PI3K-dependent Akt (Ser-473) and eNOS (Ser-1177) phosphorylation and nitric oxide-dependent cell migration, demonstrating the essential role of H-Ras. Activation of endogenous H-Ras led to recruitment and phosphorylation of eNOS at endomembranes. The loss of migratory response in cells lacking endogenous H-Ras was fully restored by modest overexpression of an endomembrane-delimited H-Ras palmitoylation mutant. These studies define a newly recognized role for endomembrane-localized H-Ras in mediating nitric oxide-dependent proangiogenic signaling.
Keywords: Angiogenesis; Cell Migration; Endomembrane; H-Ras; Nitric Oxide; Nitric-oxide Synthase; Protein Palmitoylation; Vascular Endothelial Growth Factor (VEGF).
Figures
Similar articles
-
Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells.Am J Physiol Cell Physiol. 2009 May;296(5):C1162-70. doi: 10.1152/ajpcell.00533.2008. Epub 2009 Feb 25. Am J Physiol Cell Physiol. 2009. PMID: 19244479
-
Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells.J Biol Chem. 2007 Apr 6;282(14):10660-9. doi: 10.1074/jbc.M609048200. Epub 2007 Feb 15. J Biol Chem. 2007. PMID: 17303569
-
S100A1 deficiency impairs postischemic angiogenesis via compromised proangiogenic endothelial cell function and nitric oxide synthase regulation.Circ Res. 2013 Jan 4;112(1):66-78. doi: 10.1161/CIRCRESAHA.112.275156. Epub 2012 Oct 9. Circ Res. 2013. PMID: 23048072 Free PMC article.
-
Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling.Cell Signal. 2009 Jun;21(6):906-15. doi: 10.1016/j.cellsig.2009.01.038. Cell Signal. 2009. PMID: 19385062
-
The genetics of exceptional longevity identifies new druggable targets for vascular protection and repair.Pharmacol Res. 2016 Dec;114:169-174. doi: 10.1016/j.phrs.2016.10.028. Epub 2016 Nov 3. Pharmacol Res. 2016. PMID: 27818232 Review.
Cited by
-
Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells.Antioxidants (Basel). 2019 Aug 16;8(8):315. doi: 10.3390/antiox8080315. Antioxidants (Basel). 2019. PMID: 31426416 Free PMC article. Review.
-
Curation of the Mammalian Palmitoylome Indicates a Pivotal Role for Palmitoylation in Diseases and Disorders of the Nervous System and Cancers.PLoS Comput Biol. 2015 Aug 14;11(8):e1004405. doi: 10.1371/journal.pcbi.1004405. eCollection 2015 Aug. PLoS Comput Biol. 2015. PMID: 26275289 Free PMC article.
-
Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization.J Biol Chem. 2014 Mar 21;289(12):8633-44. doi: 10.1074/jbc.M113.517219. Epub 2014 Jan 30. J Biol Chem. 2014. PMID: 24482236 Free PMC article.
-
Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation.Antioxid Redox Signal. 2020 Apr 1;32(10):677-700. doi: 10.1089/ars.2019.7963. Epub 2020 Jan 23. Antioxid Redox Signal. 2020. PMID: 31813265 Free PMC article. Review.
-
Comparative proteomic analysis of compartmentalised Ras signalling.Sci Rep. 2015 Dec 1;5:17307. doi: 10.1038/srep17307. Sci Rep. 2015. PMID: 26620772 Free PMC article.
References
-
- Hancock J. F. (2003) Ras proteins. Different signals from different locations. Nat. Rev. Mol. Cell Biol. 4, 373–384 - PubMed
-
- Swarthout J. T., Lobo S., Farh L., Croke M. R., Greentree W. K., Deschenes R. J., Linder M. E. (2005) DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J. Biol. Chem. 280, 31141–31148 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
