

PRICE: software for the targeted assembly of components of (Meta) genomic sequence data
- PMID: 23550143
- PMCID: PMC3656733
- DOI: 10.1534/g3.113.005967
PRICE: software for the targeted assembly of components of (Meta) genomic sequence data
Abstract
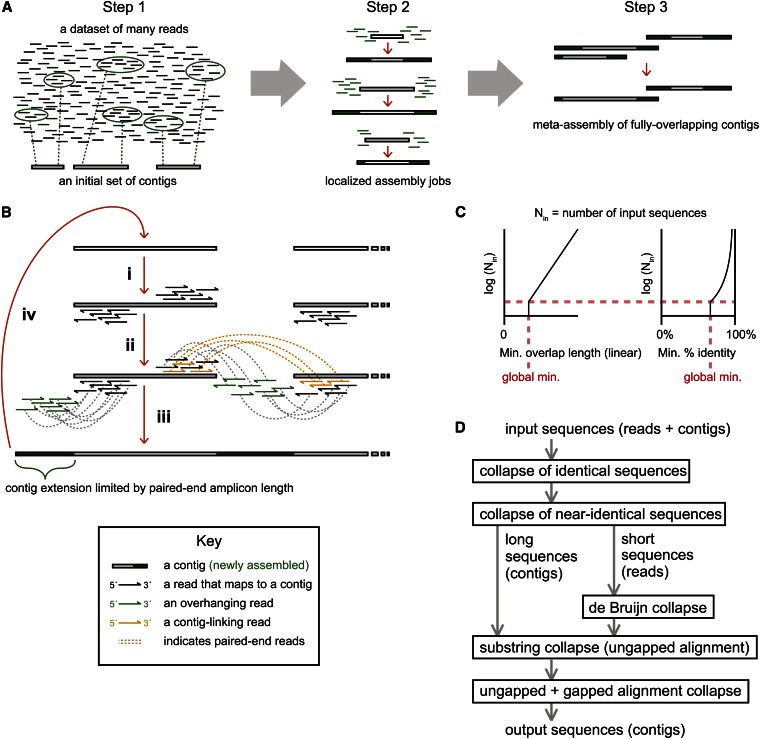
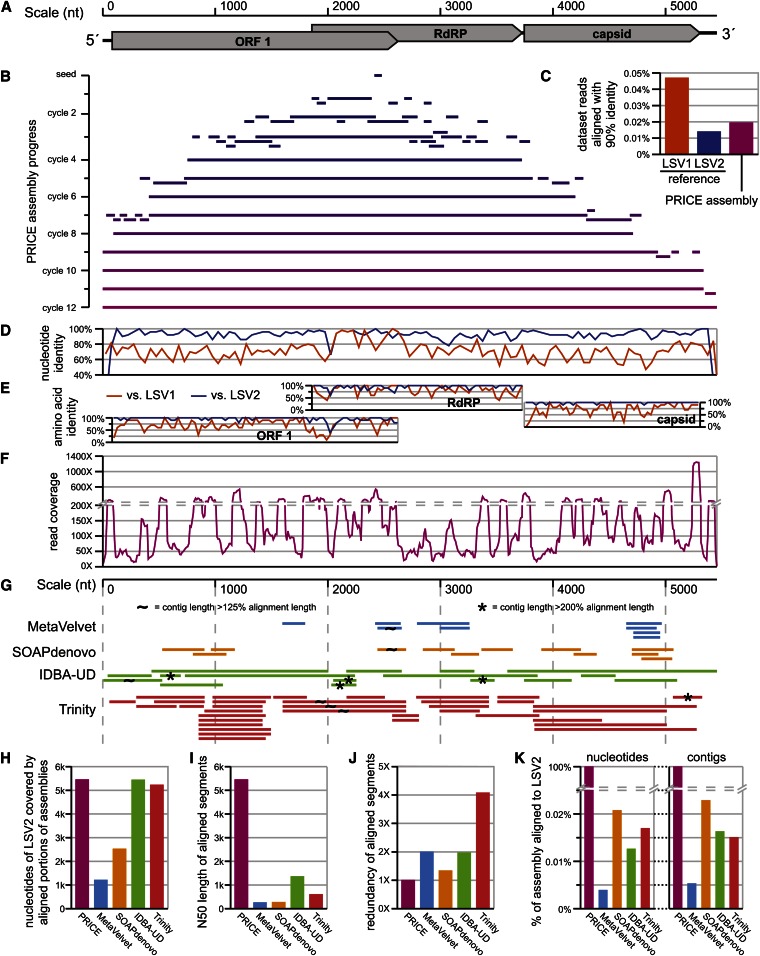
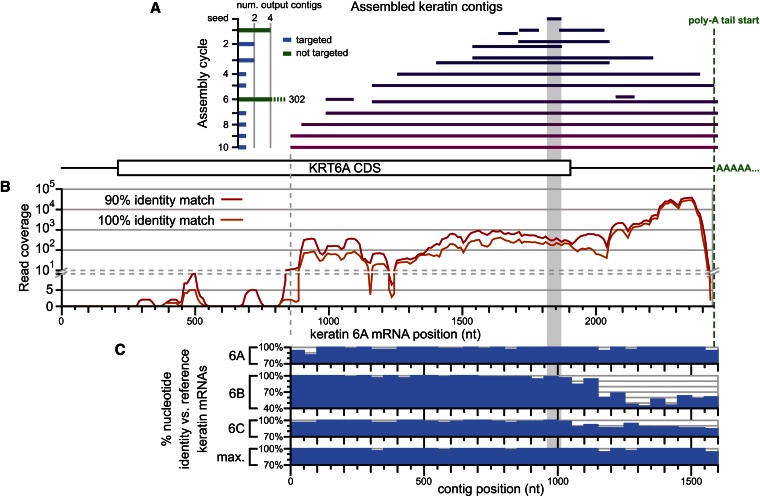
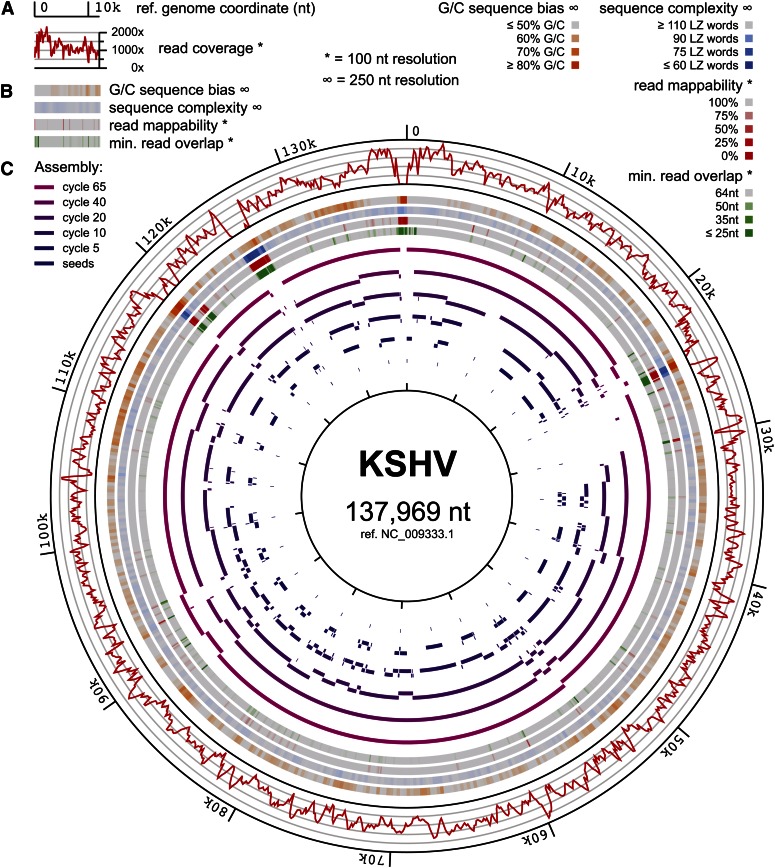
Low-cost DNA sequencing technologies have expanded the role for direct nucleic acid sequencing in the analysis of genomes, transcriptomes, and the metagenomes of whole ecosystems. Human and machine comprehension of such large datasets can be simplified via synthesis of sequence fragments into long, contiguous blocks of sequence (contigs), but most of the progress in the field of assembly has focused on genomes in isolation rather than metagenomes. Here, we present software for paired-read iterative contig extension (PRICE), a strategy for focused assembly of particular nucleic acid species using complex metagenomic data as input. We describe the assembly strategy implemented by PRICE and provide examples of its application to the sequence of particular genes, transcripts, and virus genomes from complex multicomponent datasets, including an assembly of the BCBL-1 strain of Kaposi's sarcoma-associated herpesvirus. PRICE is open-source and available for free download (derisilab.ucsf.edu/software/price/ or sourceforge.net/projects/pricedenovo/).
Keywords: KSHV; de novo genome assembly; high-throughput DNA sequencing; metagenomics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources