

Modifying muscular dystrophy through transforming growth factor-β
- PMID: 23551962
- PMCID: PMC3731412
- DOI: 10.1111/febs.12266
Modifying muscular dystrophy through transforming growth factor-β
Abstract
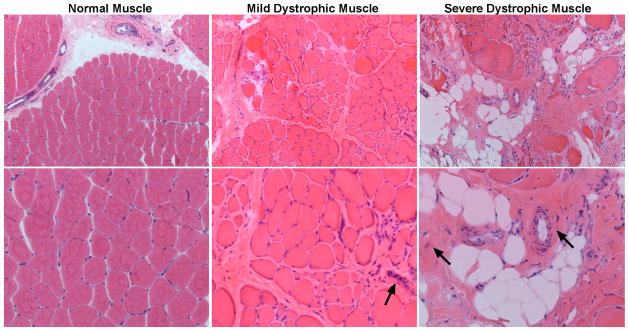
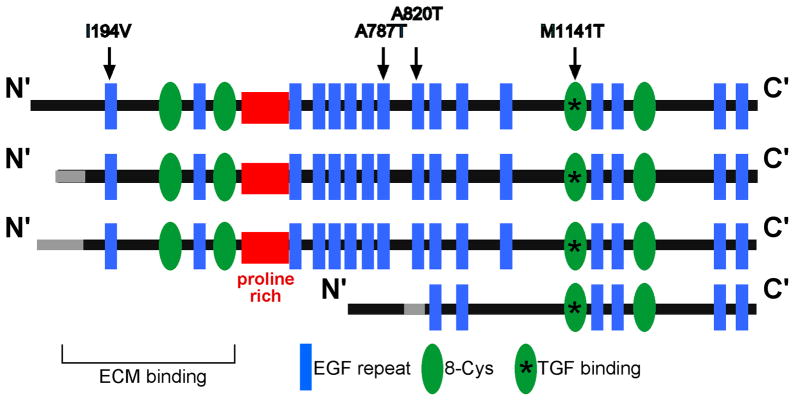
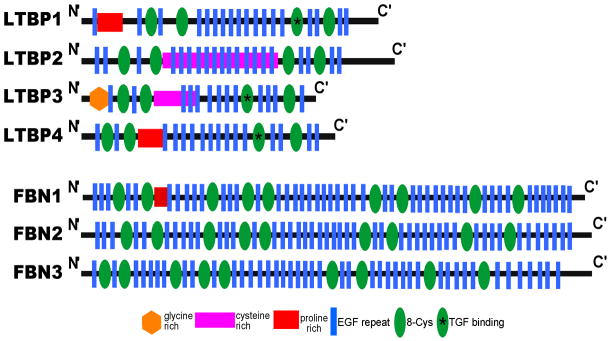
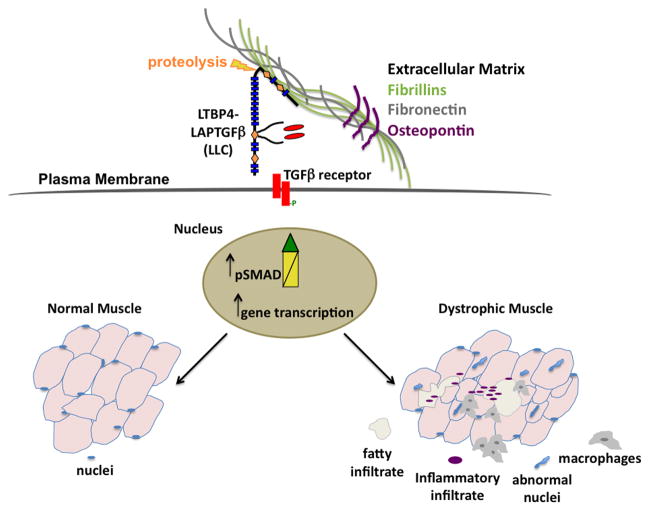
Muscular dystrophy arises from ongoing muscle degeneration and insufficient regeneration. This imbalance leads to loss of muscle, with replacement by scar or fibrotic tissue, resulting in muscle weakness and, eventually, loss of muscle function. Human muscular dystrophy is characterized by a wide range of disease severity, even when the same genetic mutation is present. This variability implies that other factors, both genetic and environmental, modify the disease outcome. There has been an ongoing effort to define the genetic and molecular bases that influence muscular dystrophy onset and progression. Modifier genes for muscle disease have been identified through both candidate gene approaches and genome-wide surveys. Multiple lines of experimental evidence have now converged on the transforming growth factor-β (TGF-β) pathway as a modifier for muscular dystrophy. TGF-β signaling is upregulated in dystrophic muscle as a result of a destabilized plasma membrane and/or an altered extracellular matrix. Given the important biological role of the TGF-β pathway, and its role beyond muscle homeostasis, we review modifier genes that alter the TGF-β pathway and approaches to modulate TGF-β activity to ameliorate muscle disease.
Keywords: extracellular matrix; genetic modifier; muscular dystrophy; transforming growth factor-β (TGF-β).
© 2013 The Authors Journal compilation © 2013 FEBS.
Figures
References
-
- Chen YW, Nagaraju K, Bakay M, McIntyre O, Rawat R, Shi R, Hoffman EP. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology. 2005;65:826–34. - PubMed
-
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–72. - PubMed
-
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, Sampson JB, Mendell JR, Wall C, King WM, Pestronk A, Florence JM, Connolly AM, Mathews KD, Stephan CM, Laubenthal KS, Wong BL, Morehart PJ, Meyer A, Finkel RS, Bonnemann CG, Medne L, Day JW, Dalton JC, Margolis MK, Hinton VJ, Weiss RB. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–66. - PMC - PubMed
-
- Flanigan KM, Ceco E, Lamar K, Kaminoh Y, Dunn DM, Mendell JR, King WM, Pestronk A, Florence JM, Mathews KD, Finkel RS, Swoboda KJ, Gappmaier E, Howard MT, Day JW, McDonald C, McNally EM, Weiss RB. LTBP4 genotype predicts age of ambulatory loss in Duchenne Muscular Dystrophy. Ann of Neurolol. 2013 in press. - PMC - PubMed
-
- Ginjaar IB, Kneppers AL, vd Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC, Weegenaar J, den Dunnen JT, Bakker E. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur J Hum Genet. 2000;8:793–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
