

Genome-wide analysis of host mRNA translation during hepatitis C virus infection
- PMID: 23552407
- PMCID: PMC3676112
- DOI: 10.1128/JVI.00538-13
Genome-wide analysis of host mRNA translation during hepatitis C virus infection
Erratum in
-
Correction for Colman et al., Genome-Wide Analysis of Host mRNA Translation during Hepatitis C Virus Infection.J Virol. 2016 May 27;90(12):5846. doi: 10.1128/JVI.00507-16. Print 2016 Jun 15. J Virol. 2016. PMID: 27234346 Free PMC article. No abstract available.
Abstract
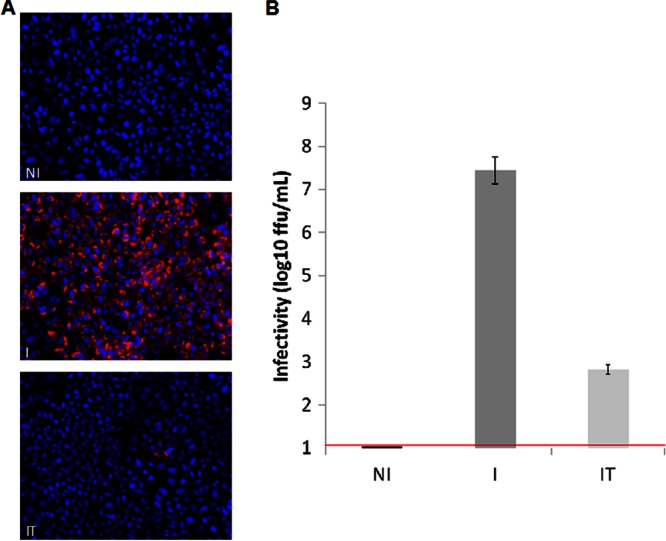
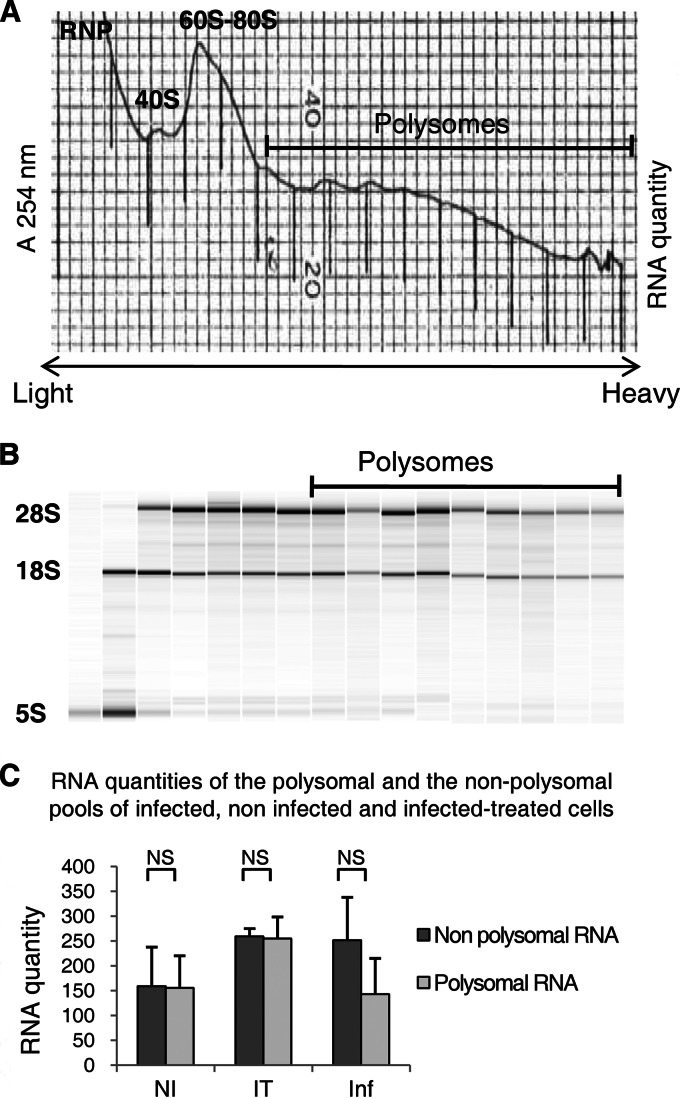
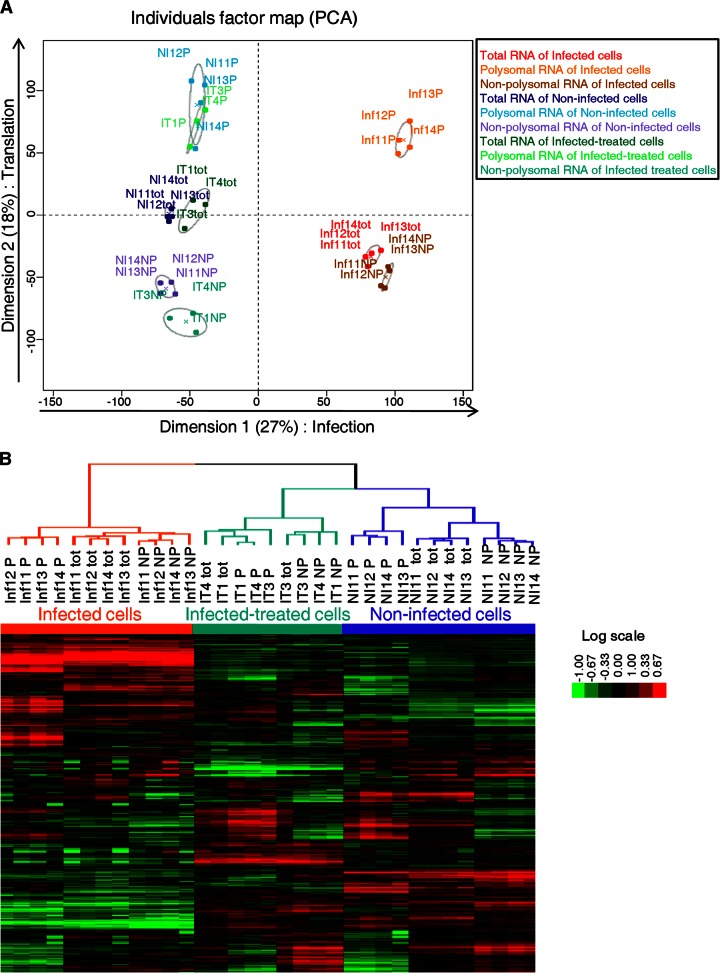
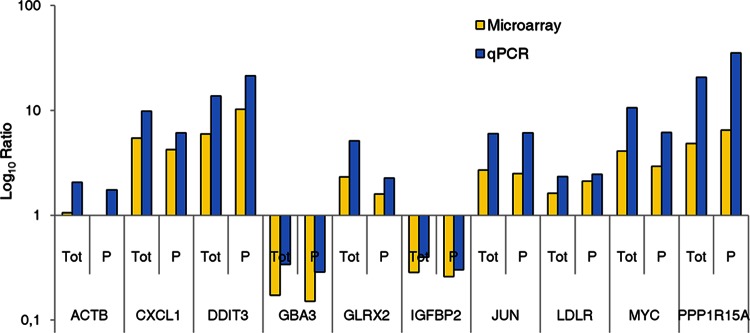
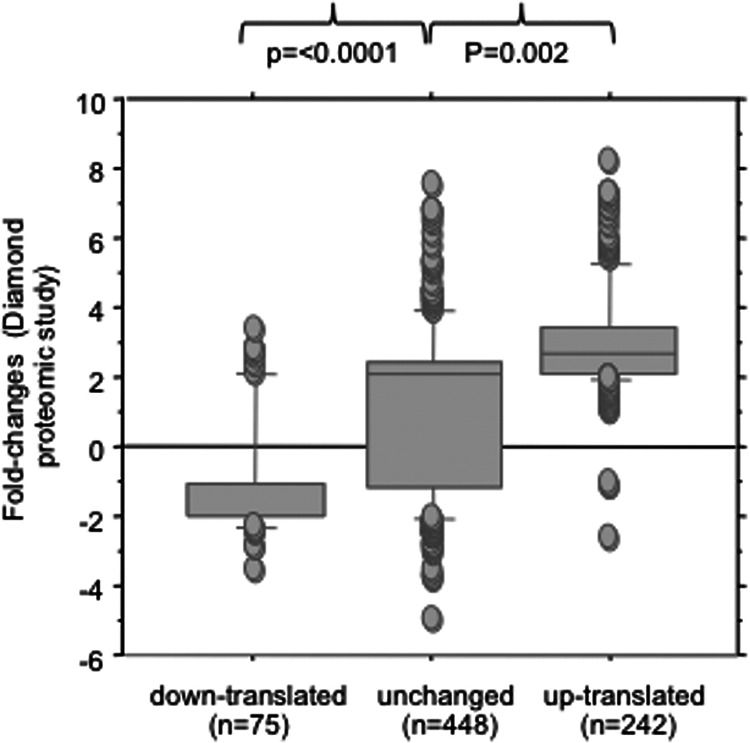
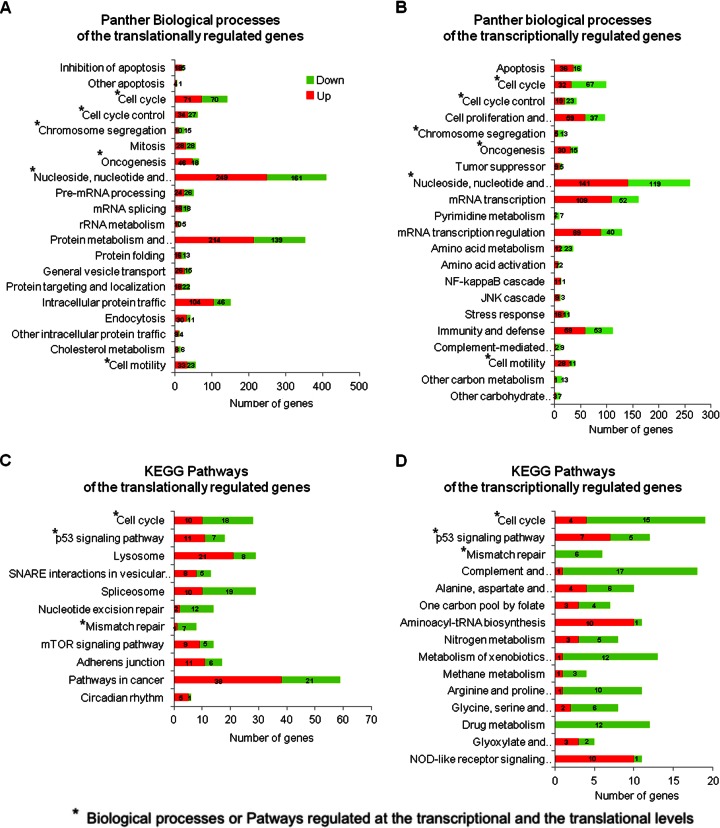
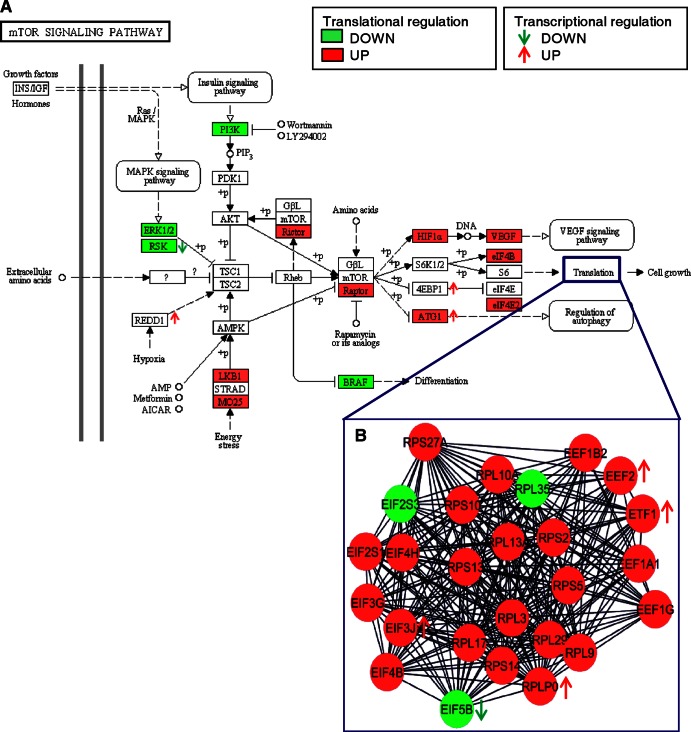
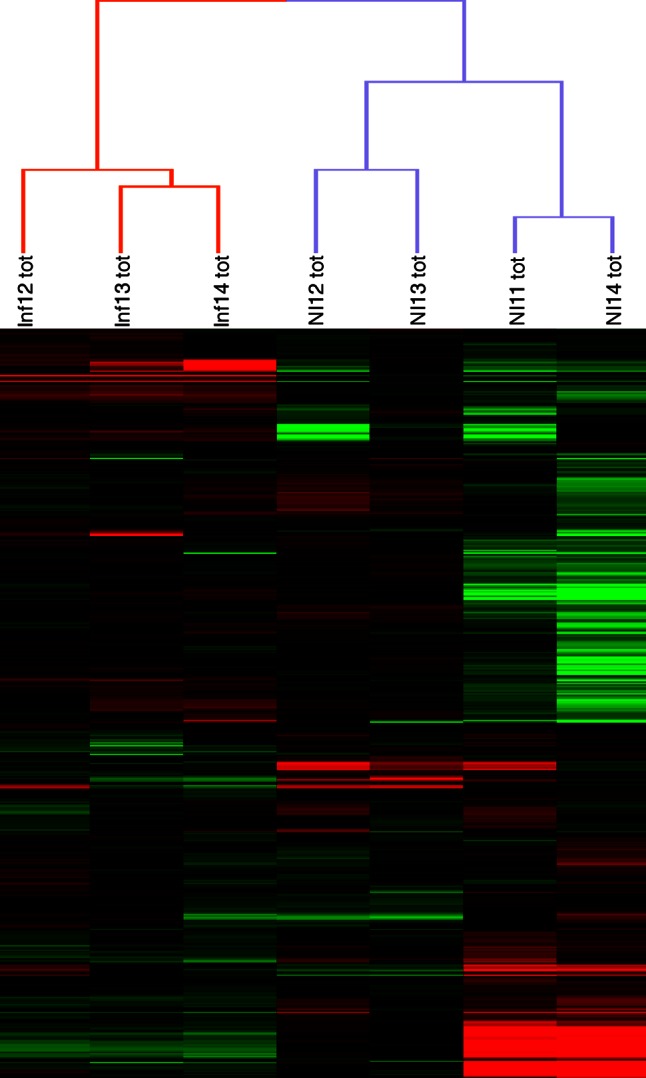
In the model of Huh-7.5.1 hepatocyte cells infected by the JFH1 hepatitis C virus (HCV) strain, transcriptomic and proteomic studies have revealed modulations of pathways governing mainly apoptosis and cell cycling. Differences between transcriptomic and proteomic studies pointed to regulations occurring at the posttranscriptional level, including the control of mRNA translation. In this study, we investigated at the genome-wide level the translational regulation occurring during HCV infection. Sucrose gradient ultracentrifugation followed by microarray analysis was used to identify translationally regulated mRNAs (mRNAs associated with ribosomes) from JFH1-infected and uninfected Huh-7.5.1 cells. Translationally regulated mRNAs were found to correspond to genes enriched in specific pathways, including vesicular transport and posttranscriptional regulation. Interestingly, the strongest translational regulation was found for mRNAs encoding proteins involved in pre-mRNA splicing, mRNA translation, and protein folding. Strikingly, these pathways were not previously identified, through transcriptomic studies, as being modulated following HCV infection. Importantly, the observed changes in host mRNA translation were directly due to HCV replication rather than to HCV entry, since they were not observed in JFH1-infected Huh-7.5.1 cells treated with a potent HCV NS3 protease inhibitor. Overall, this study highlights the need to consider, beyond transcriptomic or proteomic studies, the modulation of host mRNA translation as an important aspect of HCV infection.
Figures
References
-
- Aizaki H, Harada T, Otsuka M, Seki N, Matsuda M, Li YW, Kawakami H, Matsuura Y, Miyamura T, Suzuki T. 2002. Expression profiling of liver cell lines expressing entire or parts of hepatitis C virus open reading frame. Hepatology 36:1431–1438 - PubMed
-
- Ciccaglione AR, Marcantonio C, Tritarelli E, Tataseo P, Ferraris A, Bruni R, Dallapiccola B, Gerosolimo G, Costantino A, Rapicetta M. 2008. Microarray analysis identifies a common set of cellular genes modulated by different HCV replicon clones. BMC Genomics 9:309 doi: 10.1186/1471-2164-9-309 - DOI - PMC - PubMed
-
- Pazienza V, Clement S, Pugnale P, Conzelmann S, Pascarella S, Mangia A, Negro F. 2009. Gene expression profile of Huh-7 cells expressing hepatitis C virus genotype 1b or 3a core proteins. Liver Int. 29:661–669 - PubMed
-
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
