

Re-analysis of an original CMTX3 family using exome sequencing identifies a known BSCL2 mutation
- PMID: 23553728
- PMCID: PMC5175269
- DOI: 10.1002/mus.23743
Re-analysis of an original CMTX3 family using exome sequencing identifies a known BSCL2 mutation
Abstract
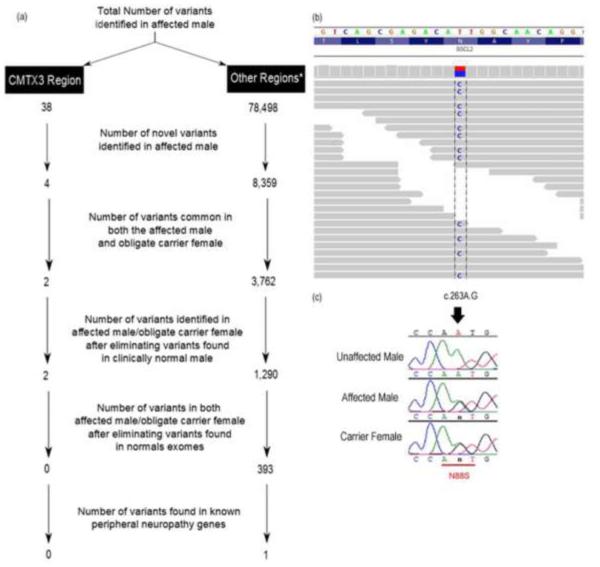
Introduction: Charcot-Marie-Tooth (CMT) disease is a group of peripheral neuropathies affecting both motor and sensory nerves. CMTX3 is an X-linked CMT locus, which maps to chromosome Xq26.3-q27.3. Initially, CMTX3 was mapped to a 31.2-Mb region in 2 American families. We have reexamined 1 of the original families (US-PED2) by next generation sequencing.
Methods: Three members of the family underwent exome sequencing. Candidate variants were validated by PCR and Sanger sequencing analysis.
Conclusion: No pathogenic coding variants localizing to the CMTX3 region were identified. However, exome sequencing identified a known BSCL2 mutation (N88S). This study demonstrates the power of exome sequencing as a tool to identify gene mutations for a small family in the absence of statistically significant linkage data.
Copyright © 2012 Wiley Periodicals, Inc.
Figures
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974;6:98–118. - PubMed
-
- Ionasescu VV. Charcot-Marie-Tooth neuropathies: from clinical description to molecular genetics. Muscle Nerve. 1995;18:267–275. - PubMed
-
- Nelis E, vanBroeckhoven C, deJonghe P, Lofgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25–33. - PubMed
-
- Dubourg O, Tardieu S, Birouk N, Gouider R, Leger JM, Maisonobe T, et al. The frequency of 17p11.2 duplication and Connexin 32 mutations in 282 Charcot-Marie-Tooth families in relation to the mode of inheritance and motor nerve conduction velocity. Neuromuscul Disord. 2001;11:458–463. - PubMed
-
- Gal A, Mucke J, Theile H, Wieacker PF, Ropers HH, Wienker TF. X-linked dominant Charcot-Marie-Tooth disease: suggestion of linkage with a cloned DNA sequence from the proximal Xq. Hum Genet. 1985;70:38–42. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
