

The p38α MAPK regulates microglial responsiveness to diffuse traumatic brain injury
- PMID: 23554495
- PMCID: PMC3667712
- DOI: 10.1523/JNEUROSCI.5399-12.2013
The p38α MAPK regulates microglial responsiveness to diffuse traumatic brain injury
Abstract
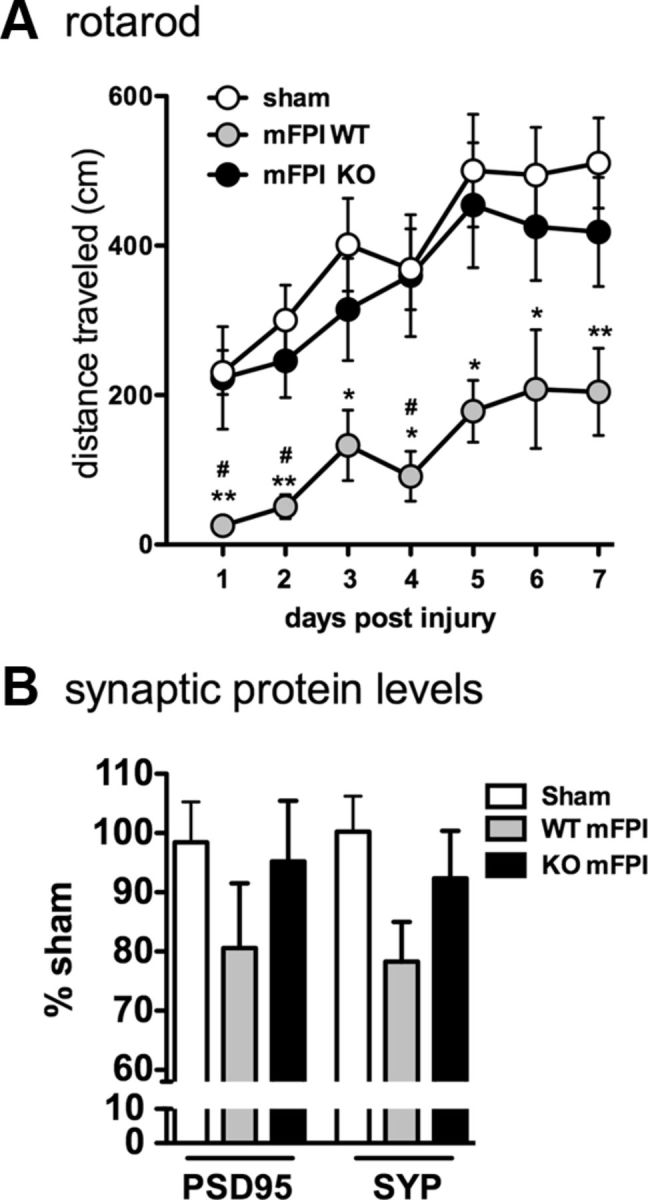
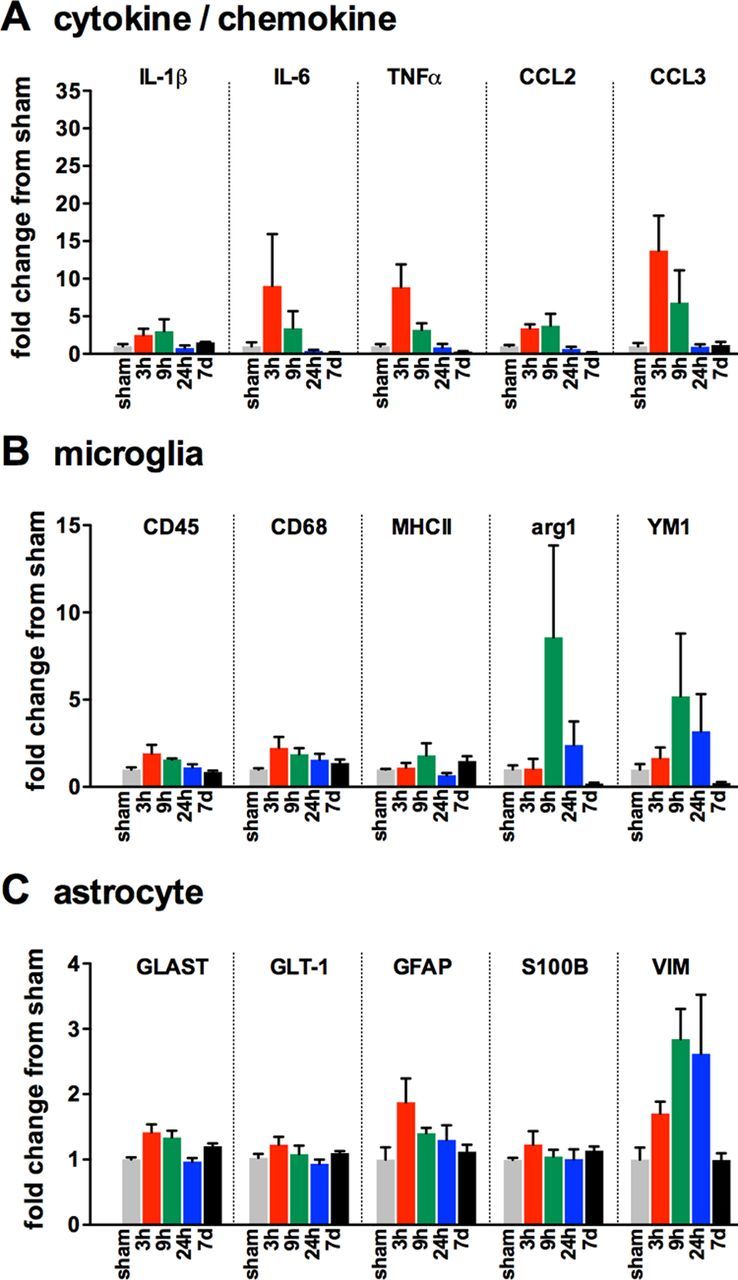
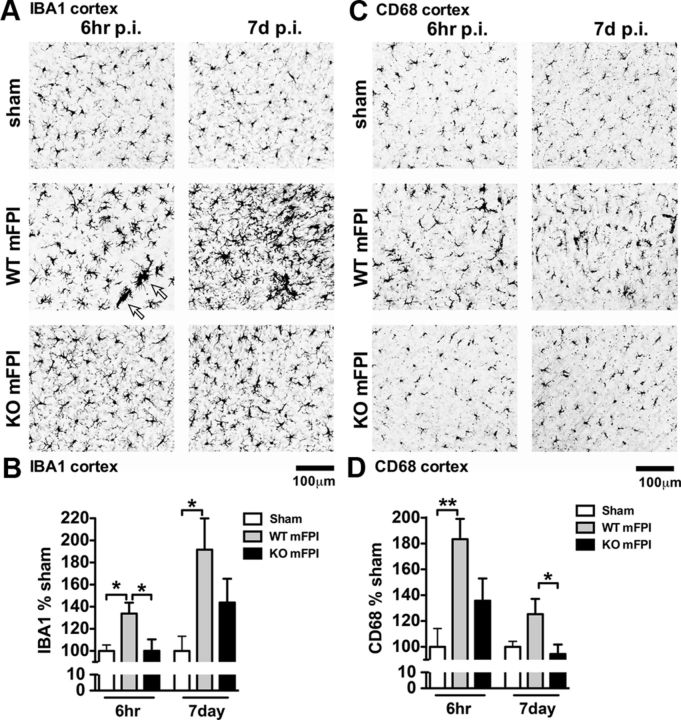
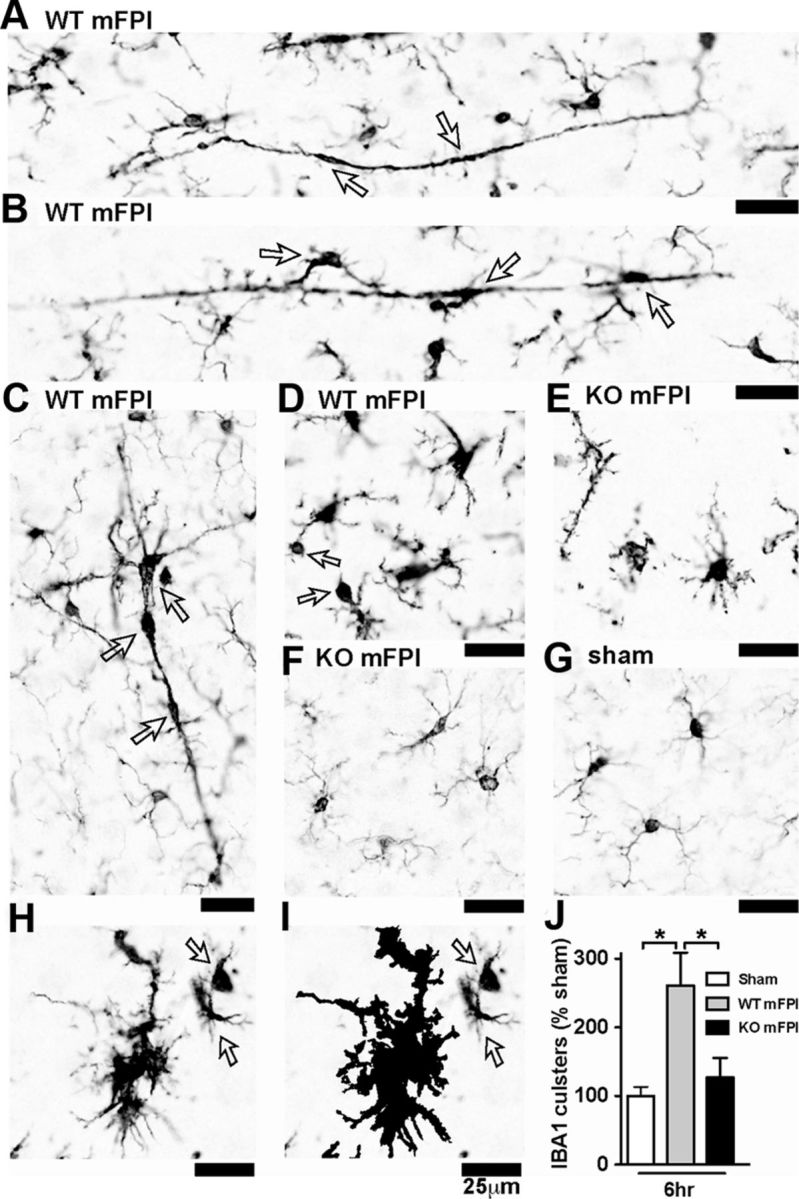
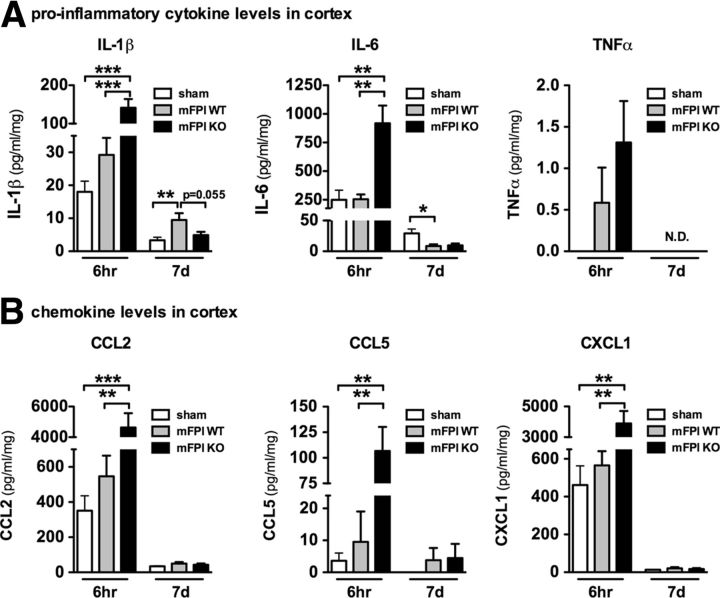
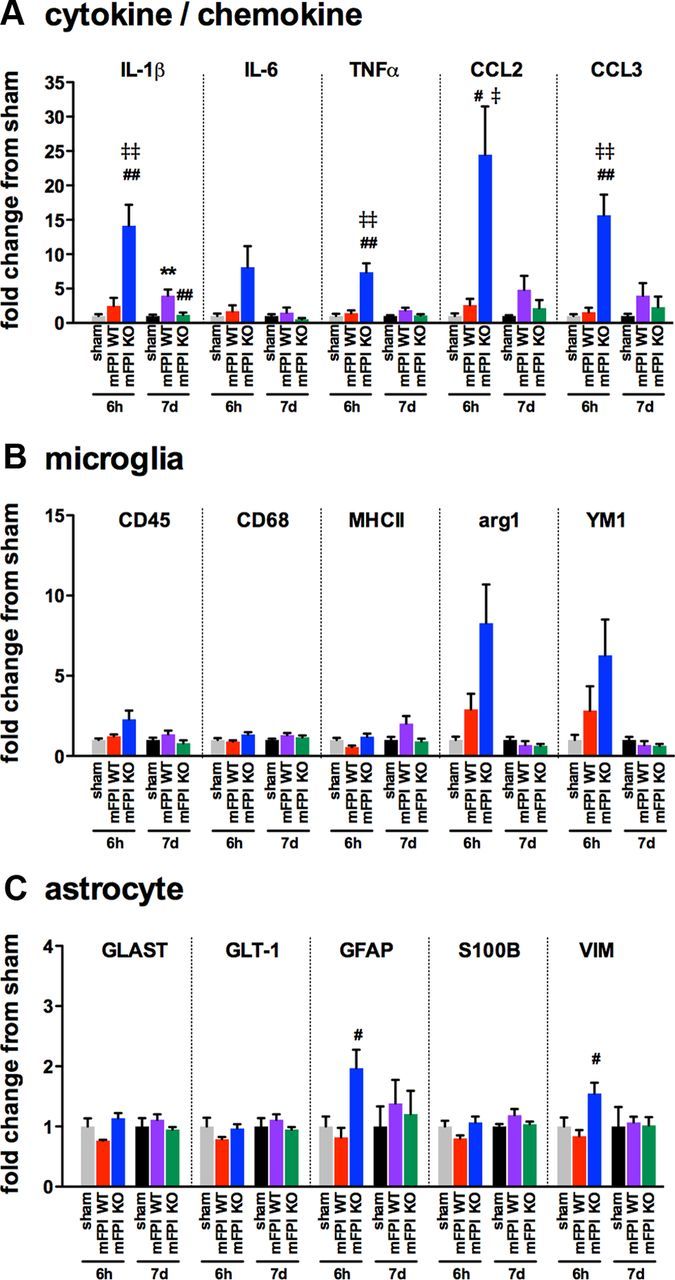
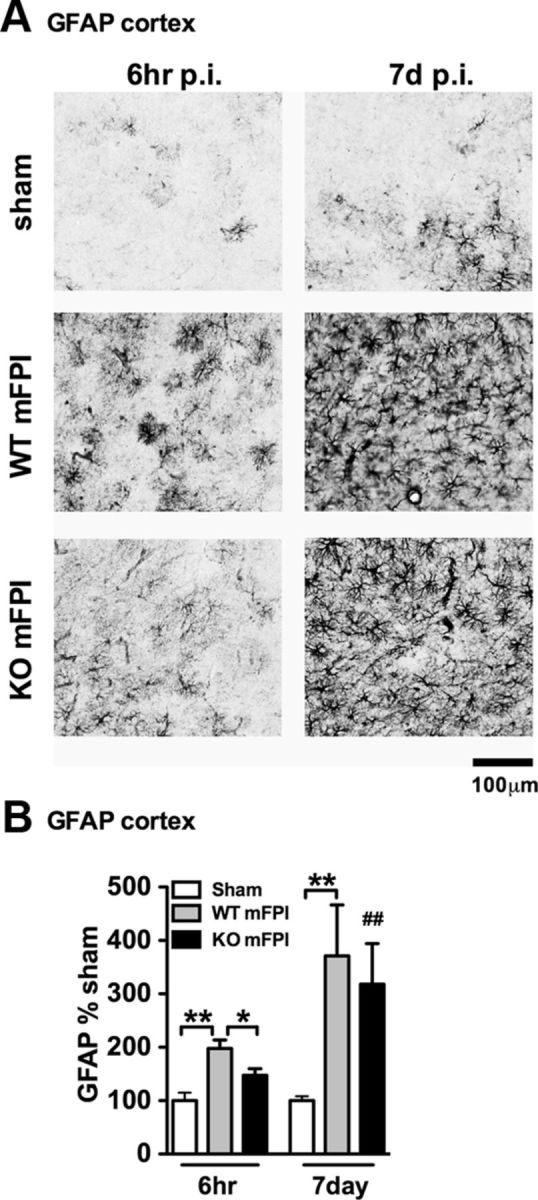
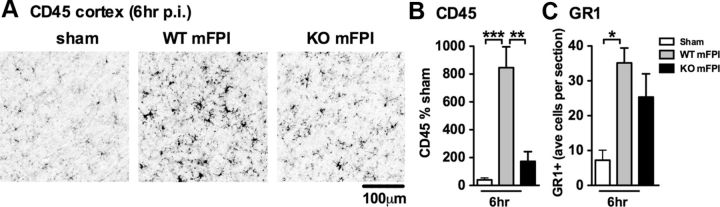
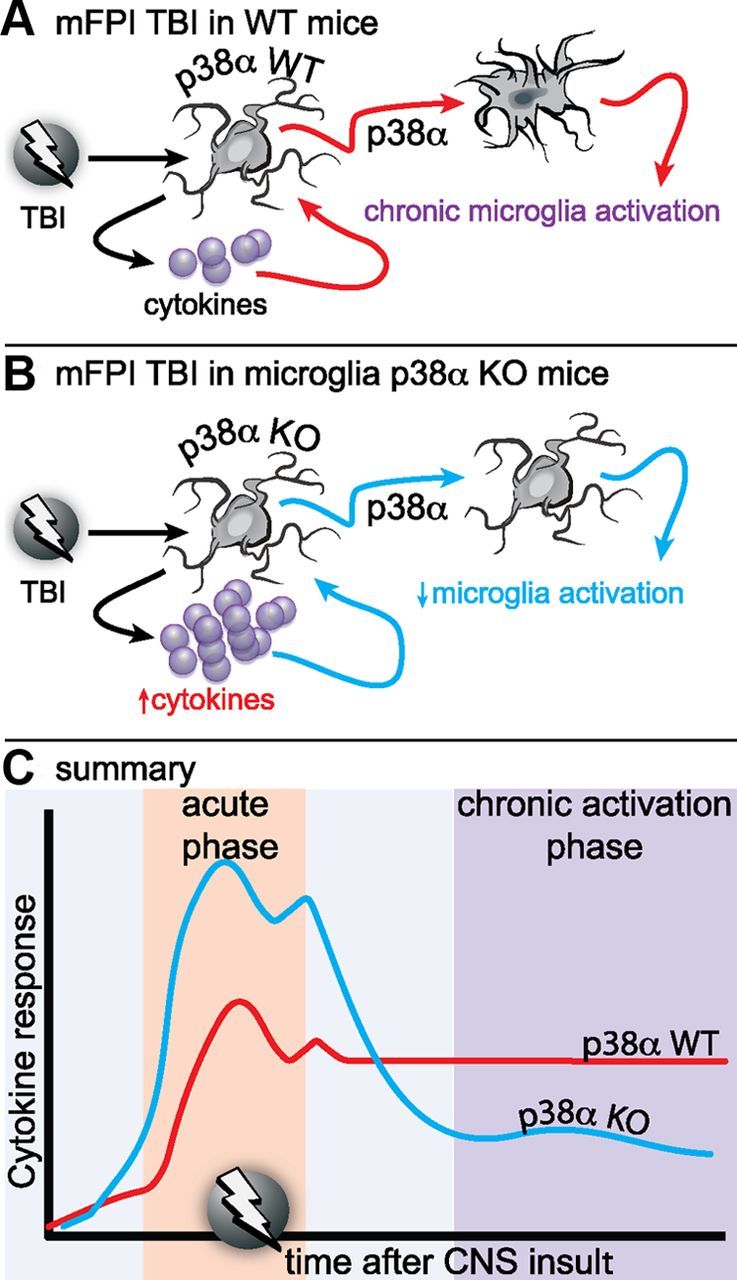
Neuropathology after traumatic brain injury (TBI) is the result of both the immediate impact injury and secondary injury mechanisms. Unresolved post-traumatic glial activation is a secondary injury mechanism that contributes to a chronic state of neuroinflammation in both animal models of TBI and human head injury patients. We recently demonstrated, using in vitro models, that p38α MAPK signaling in microglia is a key event in promoting cytokine production in response to diverse disease-relevant stressors and subsequent inflammatory neuronal dysfunction. From these findings, we hypothesized that the p38α signaling pathway in microglia could be contributing to the secondary neuropathologic sequelae after a diffuse TBI. Mice where microglia were p38α-deficient (p38α KO) were protected against TBI-induced motor deficits and synaptic protein loss. In wild-type (WT) mice, diffuse TBI produced microglia morphological activation that lasted for at least 7 d; however, p38α KO mice failed to activate this response. Unexpectedly, we found that the peak of the early, acute phase cytokine and chemokine levels was increased in injured p38α KO mice compared with injured WT mice. The increased cytokine levels in the p38α KO mice could not be accounted for by more infiltration of macrophages or neutrophils, or increased astrogliosis. By 7 d after injury, the cytokine and chemokine levels remained elevated in injured WT mice but not in p38α KO mice. Together, these data suggest that p38α balances the inflammatory response by acutely attenuating the early proinflammatory cytokine surge while perpetuating the chronic microglia activation after TBI.
Figures

References
-
- Bachstetter AD, Norris CM, Sompol P, Wilcock DM, Goulding D, Neltner JH, St Clair D, Watterson DM, Van Eldik LJ. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer's disease-related pathology. J Neurosci. 2012;32:10201–10210. - PMC - PubMed
-
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials