

Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation
- PMID: 23555611
- PMCID: PMC3608633
- DOI: 10.1371/journal.pone.0058979
Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation
Erratum in
-
Correction: Platelets Recognize Brain-Specific Glycolipid Structures, Respond to Neurovascular Damage and Promote Neuroinflammation.PLoS One. 2014 Jan 8;9(1):10.1371/annotation/0bbea8d3-1f94-48af-915c-aec02da2f5c3. doi: 10.1371/annotation/0bbea8d3-1f94-48af-915c-aec02da2f5c3. eCollection 2014. PLoS One. 2014. PMID: 29161740 Free PMC article.
Abstract
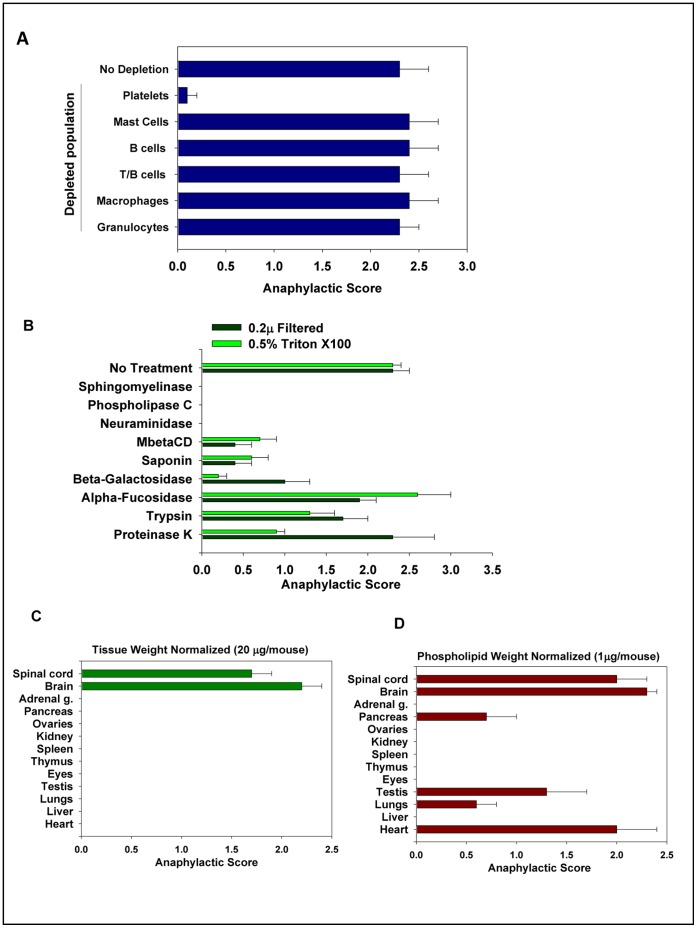
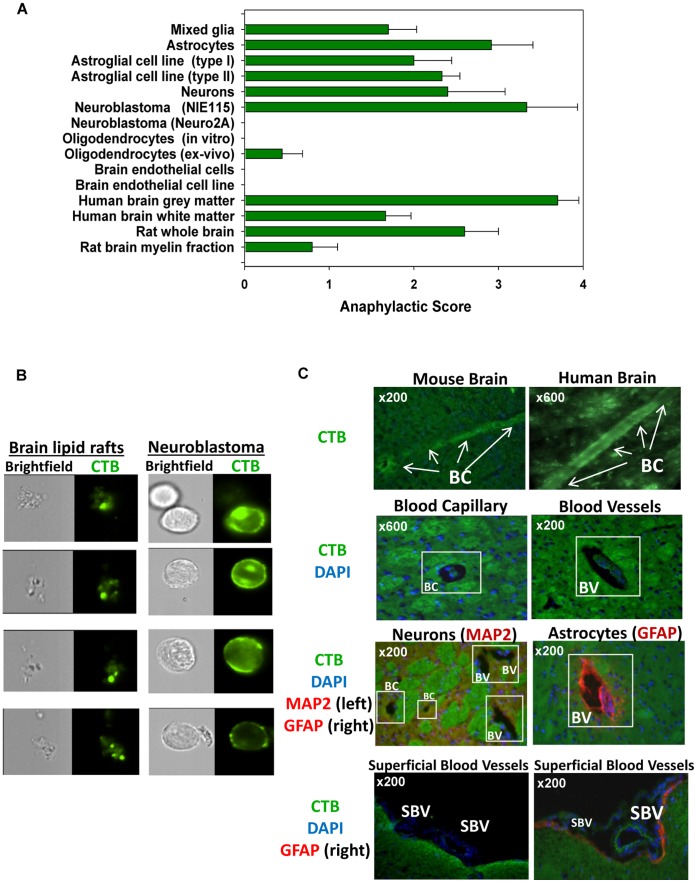
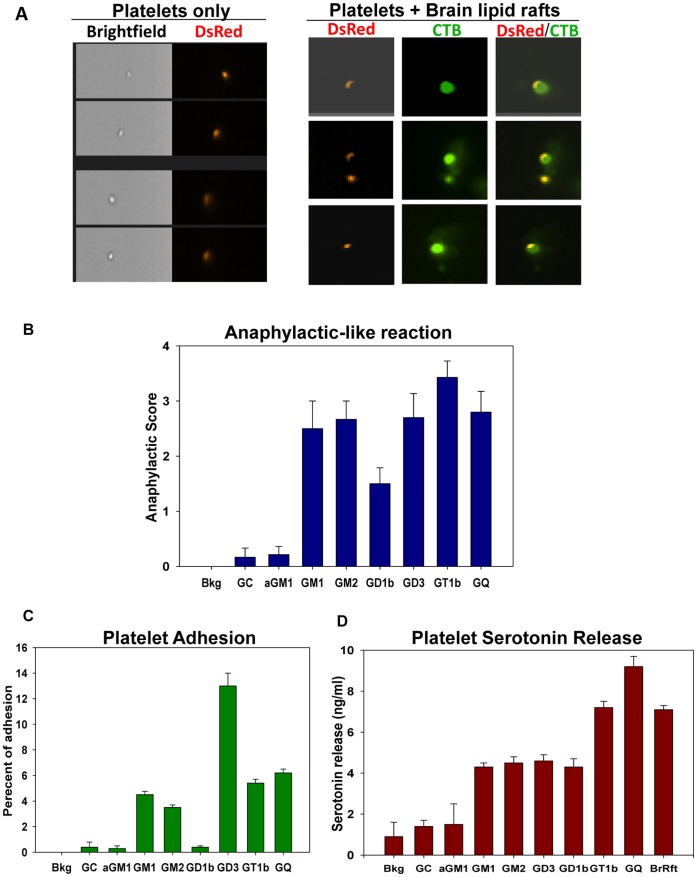
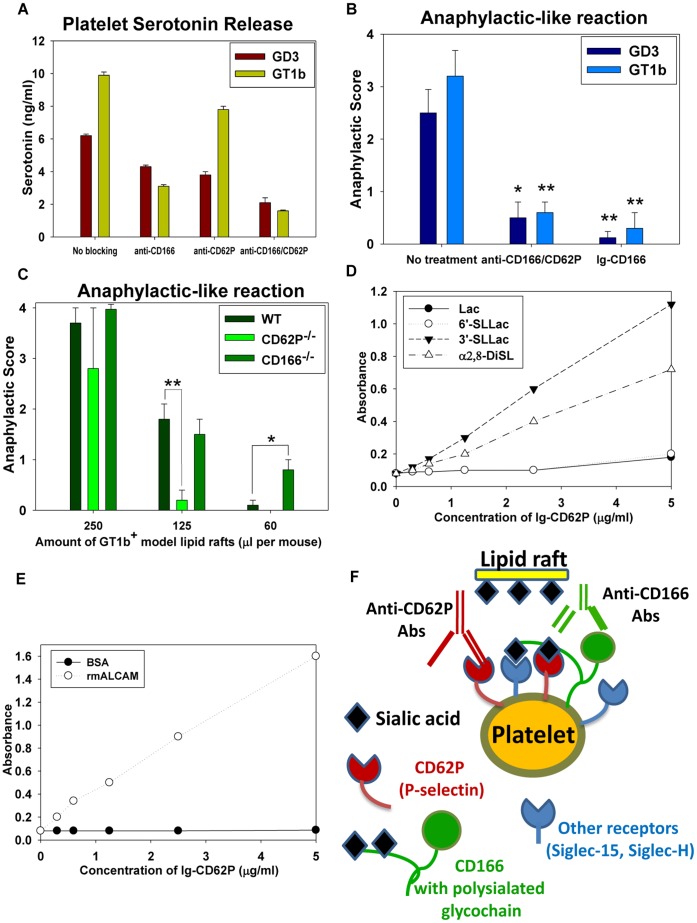
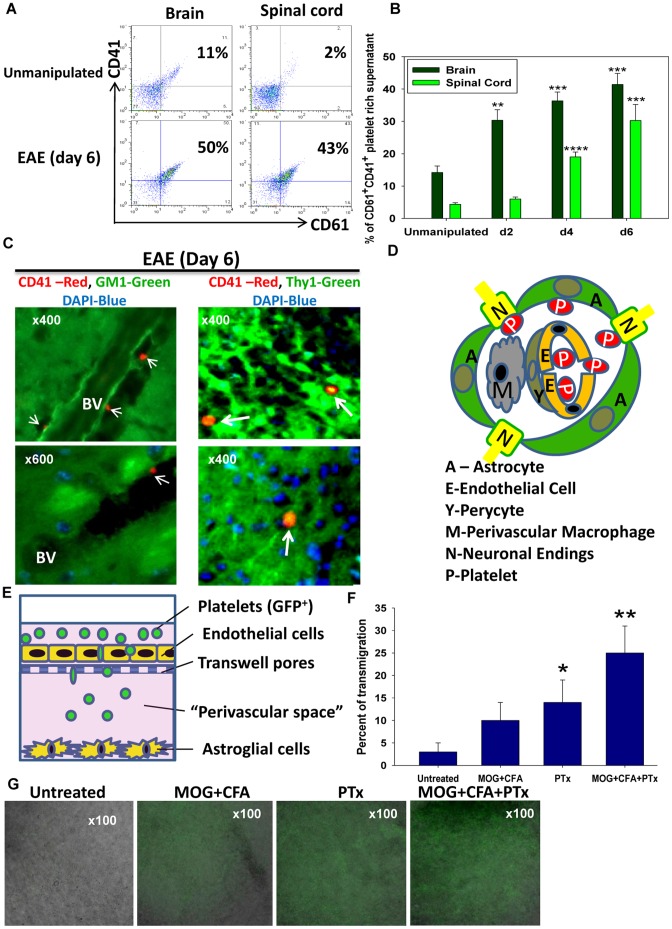
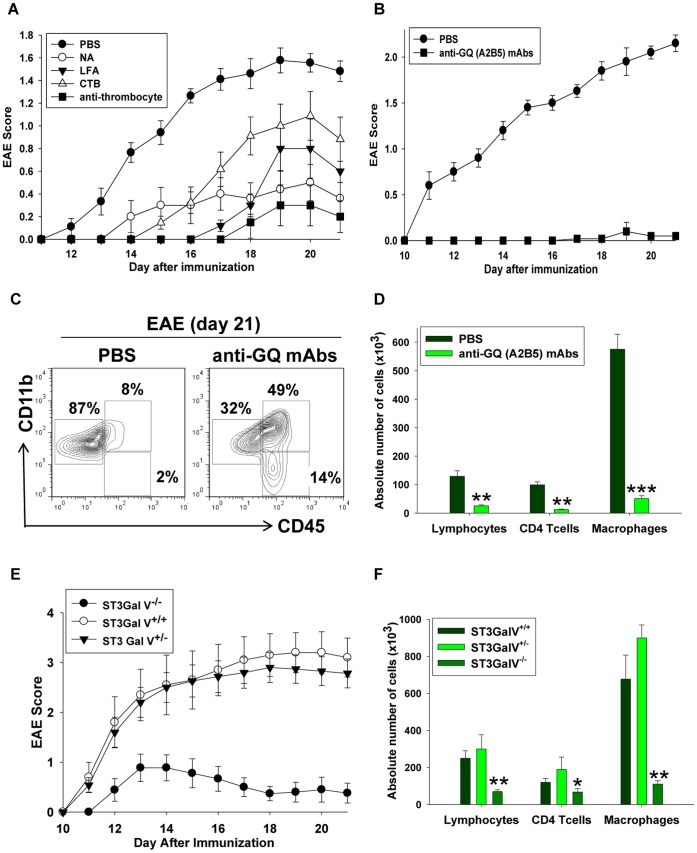
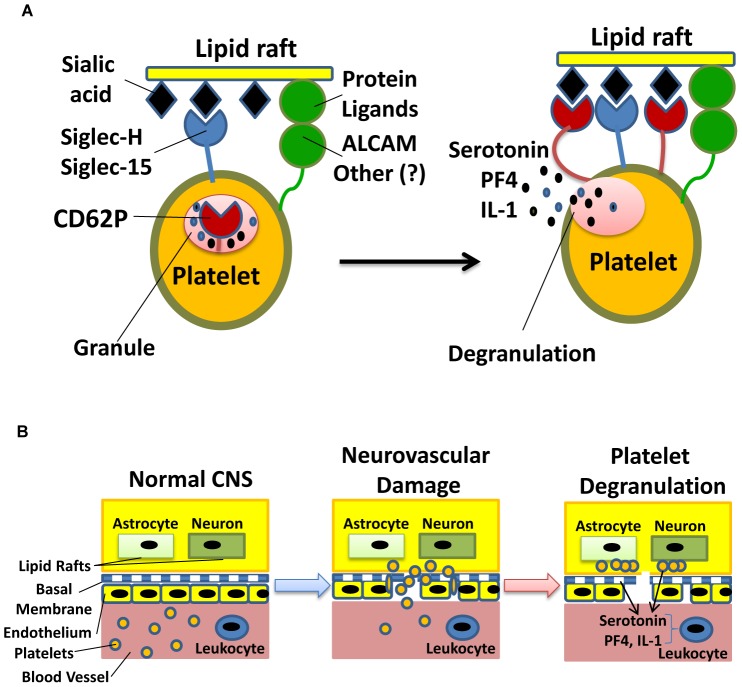
Platelets respond to vascular damage and contribute to inflammation, but their role in the neurodegenerative diseases is unknown. We found that the systemic administration of brain lipid rafts induced a massive platelet activation and degranulation resulting in a life-threatening anaphylactic-like response in mice. Platelets were engaged by the sialated glycosphingolipids (gangliosides) integrated in the rigid structures of astroglial and neuronal lipid rafts. The brain-abundant gangliosides GT1b and GQ1b were specifically recognized by the platelets and this recognition involved multiple receptors with P-selectin (CD62P) playing the central role. During the neuroinflammation, platelets accumulated in the central nervous system parenchyma, acquired an activated phenotype and secreted proinflammatory factors, thereby triggering immune response cascades. This study determines a new role of platelets which directly recognize a neuronal damage and communicate with the cells of the immune system in the pathogenesis of neurodegenerative diseases.
Conflict of interest statement
Figures
References
-
- Junt T, Schulze H, Chen Z, Massberg S, Goerge T, et al. (2007) Dynamic visualization of thrombopoiesis within bone marrow. Science 317: 1767–1770. - PubMed
-
- McNicol A, Israels SJ (2008) Beyond hemostasis: the role of platelets in inflammation, malignancy and infection. Cardiovasc Hematol Disord Drug Targets 8: 99–117. - PubMed
-
- Weyrich AS, Lindemann S, Zimmerman GA (2003) The evolving role of platelets in inflammation. J Thromb Haemost 1: 1897–1905. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
