

Gene mutation in microRNA target sites of CFTR gene: a novel pathogenetic mechanism in cystic fibrosis?
- PMID: 23555973
- PMCID: PMC3608608
- DOI: 10.1371/journal.pone.0060448
Gene mutation in microRNA target sites of CFTR gene: a novel pathogenetic mechanism in cystic fibrosis?
Abstract
Cystic fibrosis (CF) is the most frequent lethal genetic disorder among Caucasians. It depends on alterations of a chloride channel expressed by most epithelial cells and encoded by CFTR gene. Also using scanning techniques to analyze the whole coding regions of CFTR gene, mutations are not identified in up to 10% of CF alleles, and such figure increases in CFTR-related disorders (CFTR-RD). Other gene regions may be the site of causing-disease mutations. We searched for genetic variants in the 1500 bp of CFTR 3' untranslated region, typical target of microRNA (miRNA) posttranscriptional gene regulation, in either CF patients with the F508del homozygous genotype and different clinical expression (n = 20), CF (n = 32) and CFTR-RD (n = 43) patients with one or none mutation after CFTR scanning and in controls (n = 50). We identified three SNPs, one of which, the c.*1043A>C, was located in a region predicted to bind miR-433 and miR-509-3p. Such mutation was peculiar of a CFTR-RD patient that had Congenital Bilateral Absence of Vas Deferens (CBAVD), diffuse bronchiectasis, a borderline sweat chloride test and the heterozygous severe F508del mutation on the other allele. The expression analysis demonstrated that the c.*1043A>C increases the affinity for miR-509-3p and slightly decreases that for the miR-433. Both miRNAs cause in vitro a reduced expression of CFTR protein. Thus, the c.*1043A>C may act as a mild CFTR mutation enhancing the affinity for inhibitory miRNAs as a novel pathogenetic mechanism in CF.
Conflict of interest statement
Figures
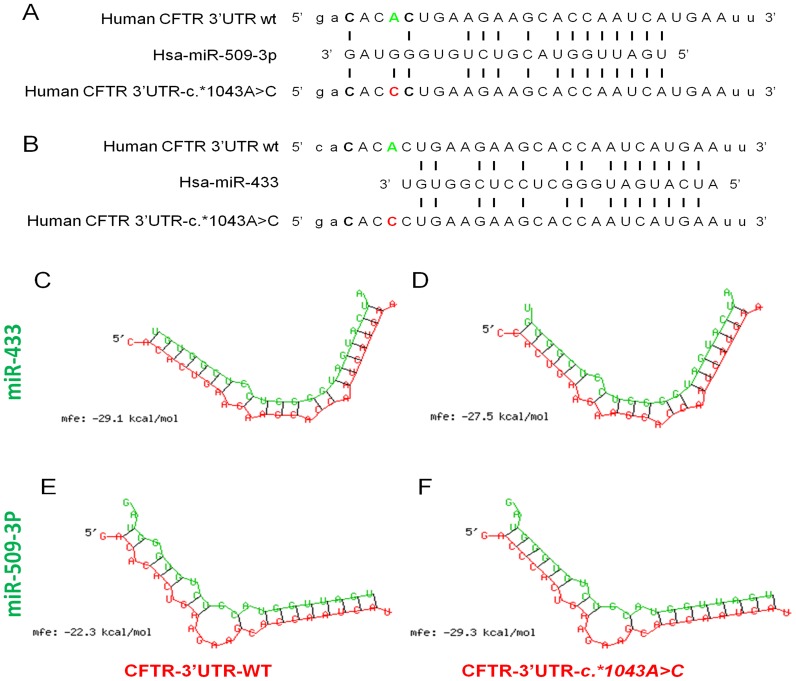
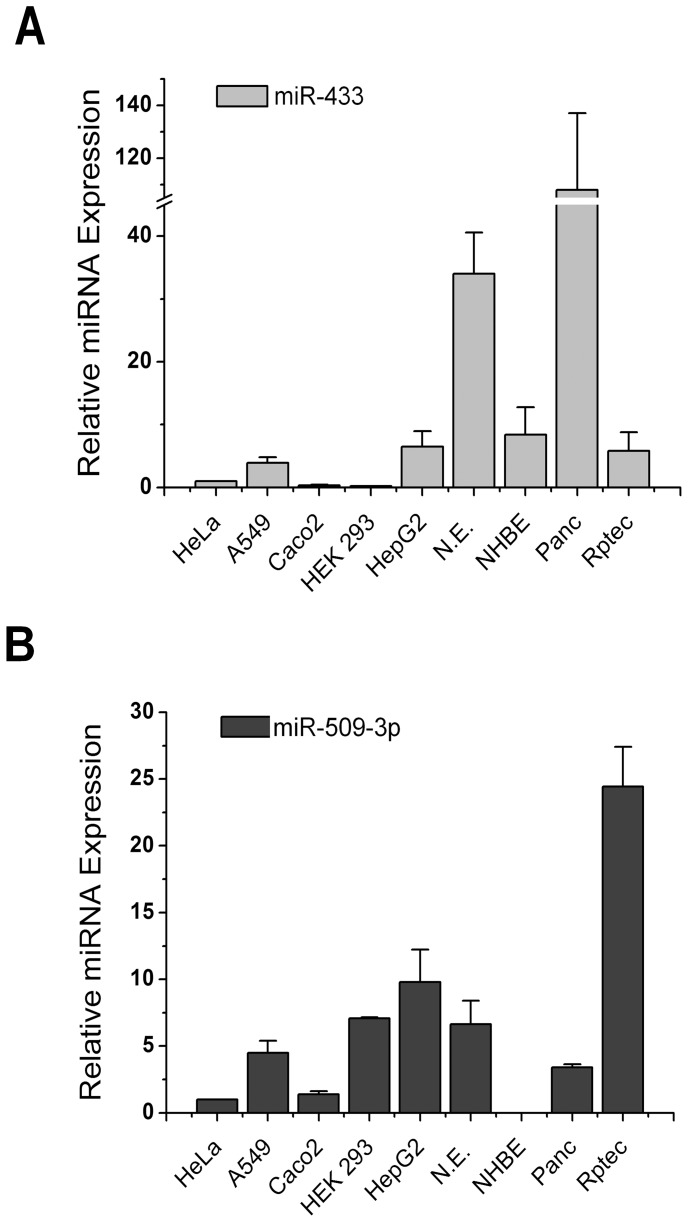
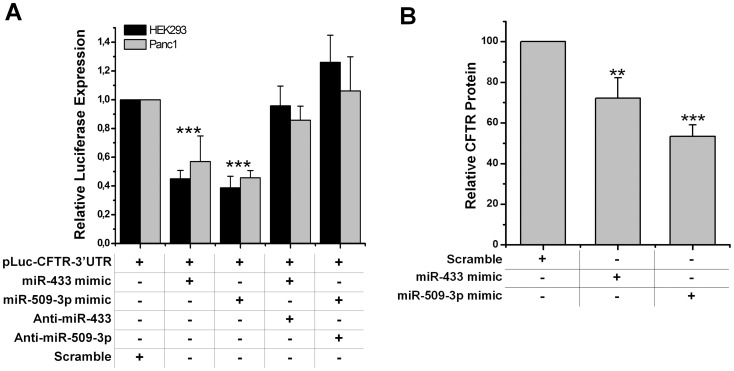
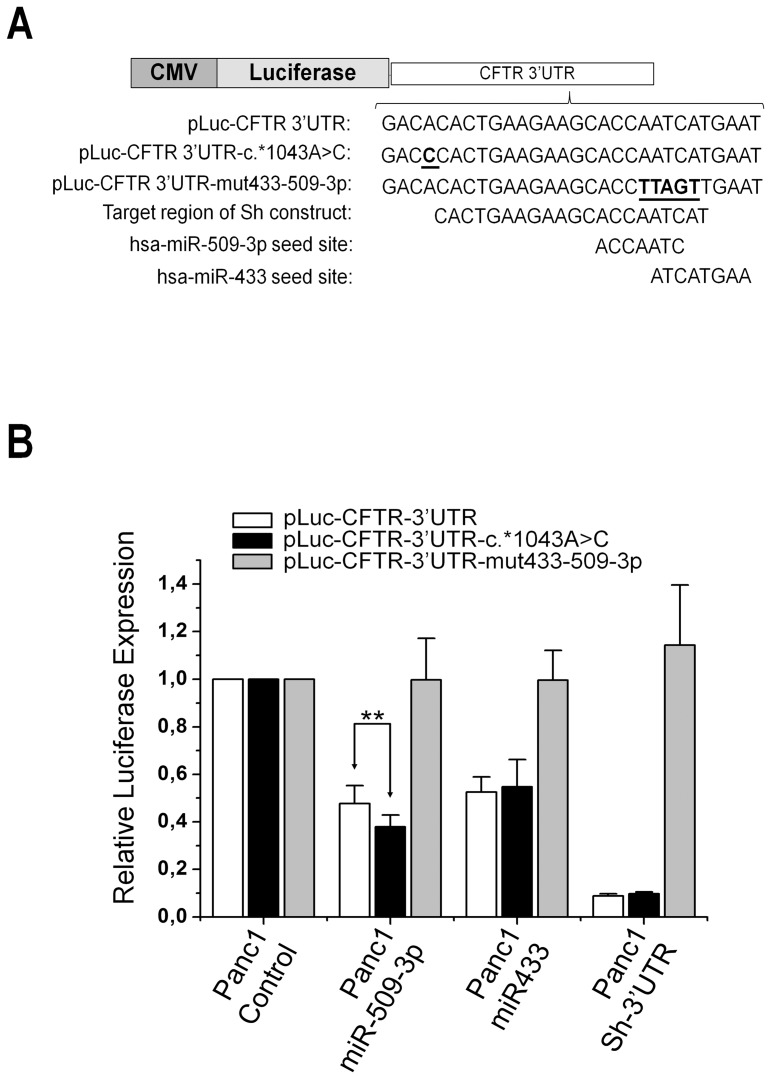
References
-
- McIntosh I, Cutting GR (1992) Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis. FASEB J 6: 2775–2782. - PubMed
-
- Castaldo G, Polizzi A, Tomaiuolo R, Cazeneuve C, Girodon E, et al. (2005) Comprehensive cystic fibrosis mutation epidemiology and haplotype characterization in a southern Italian population. Ann Hum Genet 69: 15–24. - PubMed
-
- Tomaiuolo R, Sangiuolo F, Bombieri C, Bonizzato A, Cardillo G, et al. (2008) Epidemiology and a novel procedure for large scale analysis of CFTR rearrangements in classic and atypical CF patients: a multicentric Italian study. J Cyst Fibros 7: 347–351. - PubMed
-
- Amato F, Bellia C, Cardillo G, Castaldo G, Ciaccio M, et al. (2011) Extensive molecular analysis of patients bearing CFTR-related disorders. J Mol Diagn 14: 81–89. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
