

Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways
- PMID: 23562274
- PMCID: PMC5691327
- DOI: 10.1016/j.ajpath.2013.02.008
Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways
Abstract
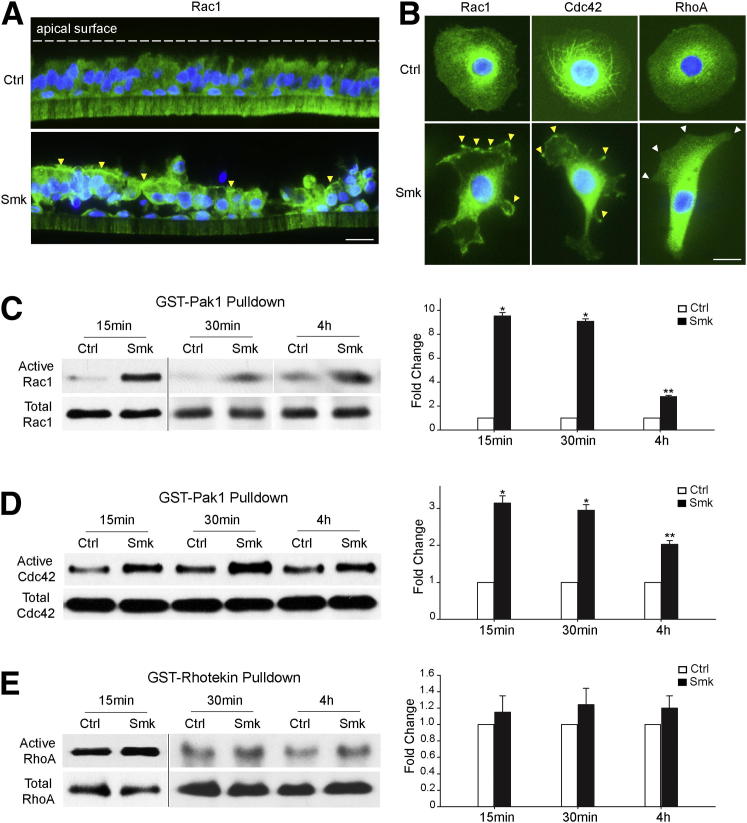
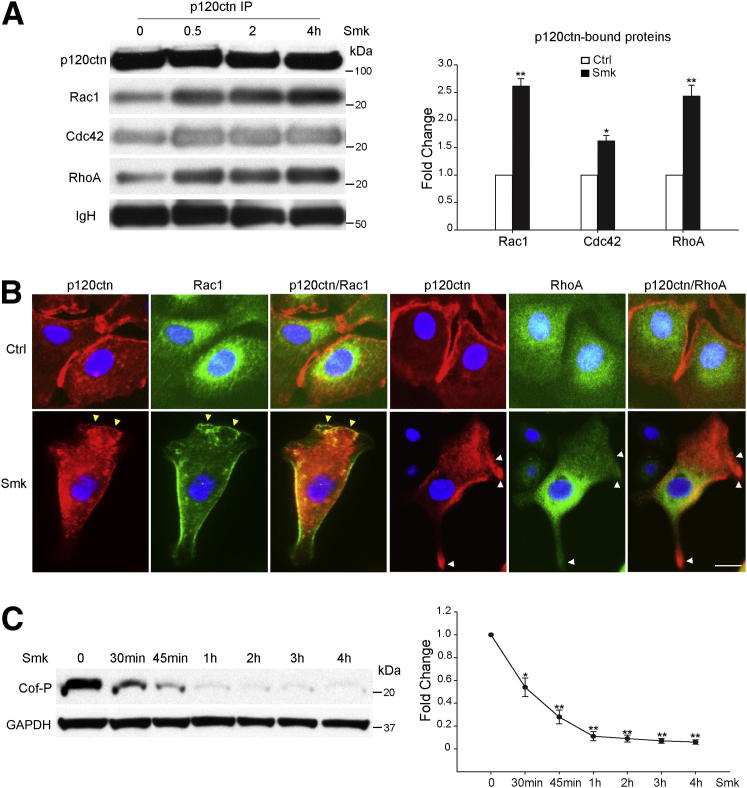
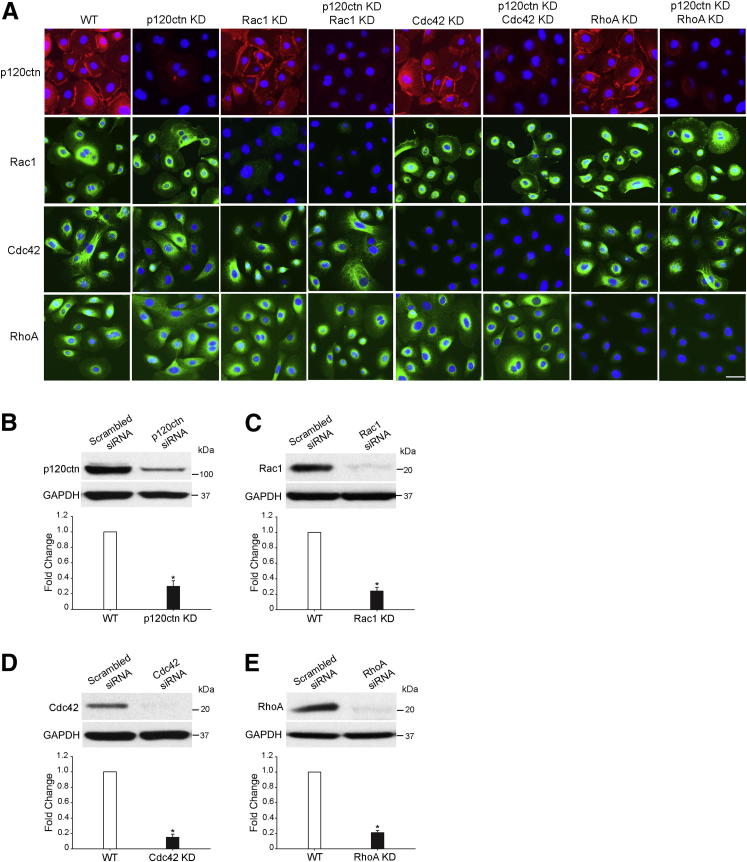
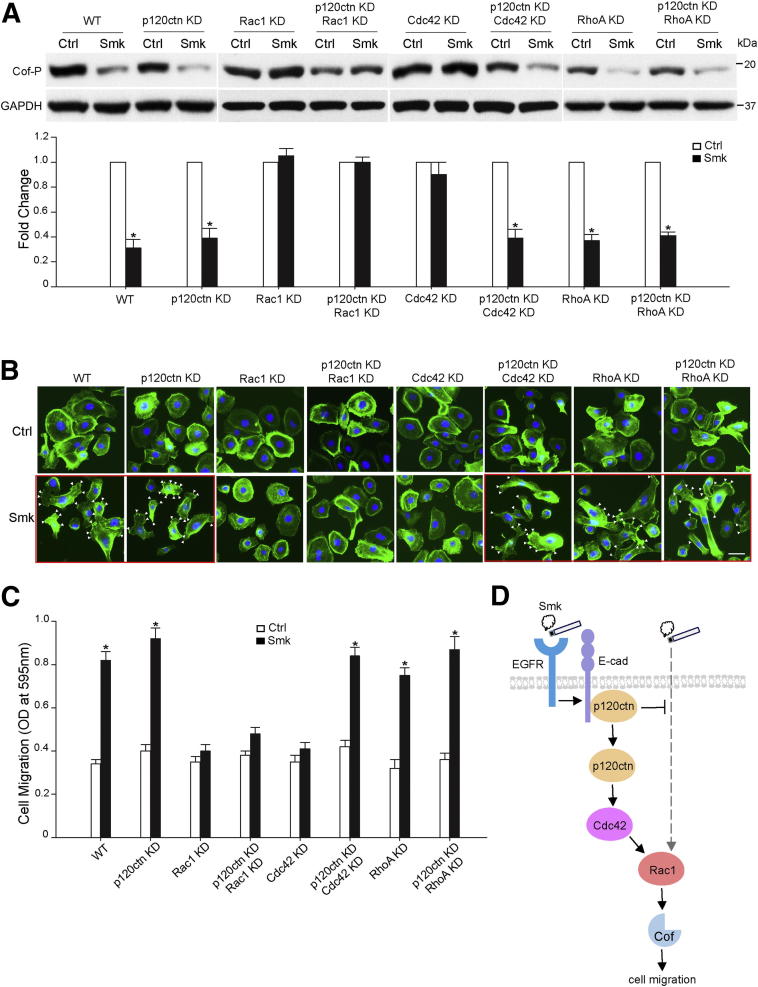
The adherens junction protein p120-catenin (p120ctn) shuttles between E-cadherin-bound and cytoplasmic pools to regulate E-cadherin/catenin complex stability and cell migration, respectively. When released from the adherens junction, p120ctn promotes cell migration through modulation of the Rho GTPases Rac1, Cdc42, and RhoA. Accordingly, the down-regulation and cytoplasmic mislocalization of p120ctn has been reported in all subtypes of lung cancers and is associated with grave prognosis. Previously, we reported that cigarette smoke induced cytoplasmic translocation of p120ctn and cell migration, but the underlying mechanism was unclear. Using primary human bronchial epithelial cells exposed to smoke-concentrated medium (Smk), we observed the translocation of Rac1 and Cdc42, but not RhoA, to the leading edge of polarized and migrating human bronchial epithelial cells. Rac1 and Cdc42 were robustly activated by smoke, whereas RhoA was inhibited. Accordingly, siRNA knockdown of Rac1 or Cdc42 completely abolished Smk-induced cell migration, whereas knockdown of RhoA had no effect. p120ctn/Rac1 double knockdown completely abolished Smk-induced cell migration, whereas p120ctn/Cdc42 double knockdown did not. These data suggested that Rac1 and Cdc42 coactivation was essential to smoke-promoted cell migration in the presence of p120ctn, whereas migration proceeded via Rac1 alone in the absence of p120ctn. Thus, Rac1 may provide an omnipotent therapeutic target in reversing cell migration during the early (intact p120ctn) and late (loss of p120ctn) stages of lung carcinogenesis.
Copyright © 2013 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
p120 catenin regulates the actin cytoskeleton via Rho family GTPases.J Cell Biol. 2000 Aug 7;150(3):567-80. doi: 10.1083/jcb.150.3.567. J Cell Biol. 2000. PMID: 10931868 Free PMC article.
-
Cigarette smoke disrupts the integrity of airway adherens junctions through the aberrant interaction of p120-catenin with the cytoplasmic tail of MUC1.J Pathol. 2013 Jan;229(1):74-86. doi: 10.1002/path.4070. Epub 2012 Sep 28. J Pathol. 2013. PMID: 22833523 Free PMC article.
-
p120-catenin modulates airway epithelial cell migration induced by cigarette smoke.Biochem Biophys Res Commun. 2012 Jan 6;417(1):49-55. doi: 10.1016/j.bbrc.2011.11.048. Epub 2011 Nov 19. Biochem Biophys Res Commun. 2012. PMID: 22120634 Free PMC article.
-
Regulation of Rho family GTPases by cell-cell and cell-matrix adhesion.Biol Res. 2002;35(2):239-46. doi: 10.4067/s0716-97602002000200016. Biol Res. 2002. PMID: 12415742 Review.
-
Functions of p120ctn isoforms in cell-cell adhesion and intracellular signaling.Front Biosci (Landmark Ed). 2012 Jan 1;17(5):1669-94. doi: 10.2741/4012. Front Biosci (Landmark Ed). 2012. PMID: 22201829 Review.
Cited by
-
Gene expression profile of human lung epithelial cells chronically exposed to single-walled carbon nanotubes.Nanoscale Res Lett. 2015 Jan 27;10:12. doi: 10.1186/s11671-014-0707-0. eCollection 2015. Nanoscale Res Lett. 2015. PMID: 25852310 Free PMC article.
-
Anchored PDE4 regulates chloride conductance in wild-type and ΔF508-CFTR human airway epithelia.FASEB J. 2014 Feb;28(2):791-801. doi: 10.1096/fj.13-240861. Epub 2013 Nov 7. FASEB J. 2014. PMID: 24200884 Free PMC article.
-
Knockdown of RhoQ, a member of Rho GTPase, accelerates TGF-β-induced EMT in human lung adenocarcinoma.Biochem Biophys Rep. 2022 Sep 11;32:101346. doi: 10.1016/j.bbrep.2022.101346. eCollection 2022 Dec. Biochem Biophys Rep. 2022. PMID: 36120491 Free PMC article.
-
Role of protein kinase N2 (PKN2) in cigarette smoke-mediated oncogenic transformation of oral cells.J Cell Commun Signal. 2018 Dec;12(4):709-721. doi: 10.1007/s12079-017-0442-2. Epub 2018 Feb 26. J Cell Commun Signal. 2018. PMID: 29480433 Free PMC article.
-
Soluble Wood Smoke Extract Promotes Barrier Dysfunction in Alveolar Epithelial Cells through a MAPK Signaling Pathway.Sci Rep. 2019 Jul 11;9(1):10027. doi: 10.1038/s41598-019-46400-8. Sci Rep. 2019. PMID: 31296909 Free PMC article.
References
-
- Proctor R.N. The global smoking epidemic: a history and status report. Clin Lung Cancer. 2004;5:371–376. - PubMed
-
- Dasari V., Gallup M., Lemjabbar H., Maltseva I., McNamara N. Epithelial-mesenchymal transition in lung cancer: is tobacco the “smoking gun”? Am J Respir Cell Mol Biol. 2006;35:3–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
