

Metagenomic study of the viruses of African straw-coloured fruit bats: detection of a chiropteran poxvirus and isolation of a novel adenovirus
- PMID: 23562481
- PMCID: PMC3667569
- DOI: 10.1016/j.virol.2013.03.014
Metagenomic study of the viruses of African straw-coloured fruit bats: detection of a chiropteran poxvirus and isolation of a novel adenovirus
Abstract
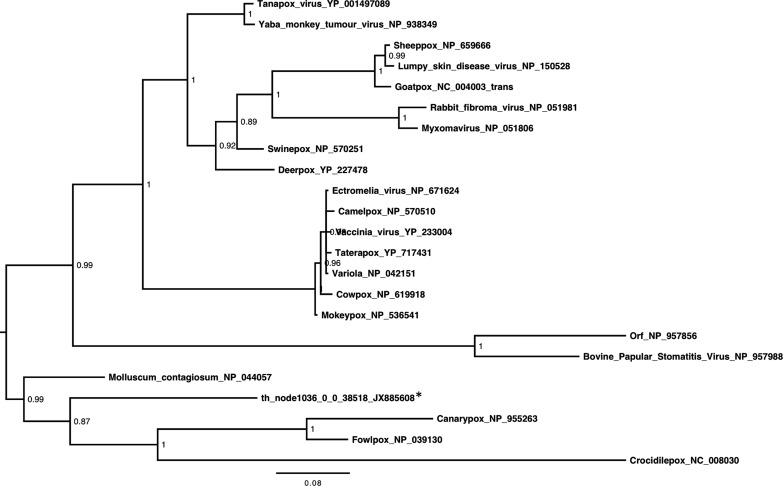
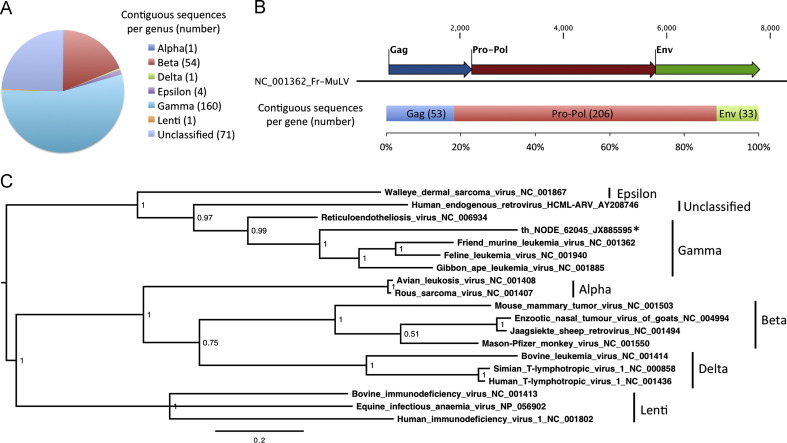
Viral emergence as a result of zoonotic transmission constitutes a continuous public health threat. Emerging viruses such as SARS coronavirus, hantaviruses and henipaviruses have wildlife reservoirs. Characterising the viruses of candidate reservoir species in geographical hot spots for viral emergence is a sensible approach to develop tools to predict, prevent, or contain emergence events. Here, we explore the viruses of Eidolon helvum, an Old World fruit bat species widely distributed in Africa that lives in close proximity to humans. We identified a great abundance and diversity of novel herpes and papillomaviruses, described the isolation of a novel adenovirus, and detected, for the first time, sequences of a chiropteran poxvirus closely related with Molluscum contagiosum. In sum, E. helvum display a wide variety of mammalian viruses, some of them genetically similar to known human pathogens, highlighting the possibility of zoonotic transmission.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
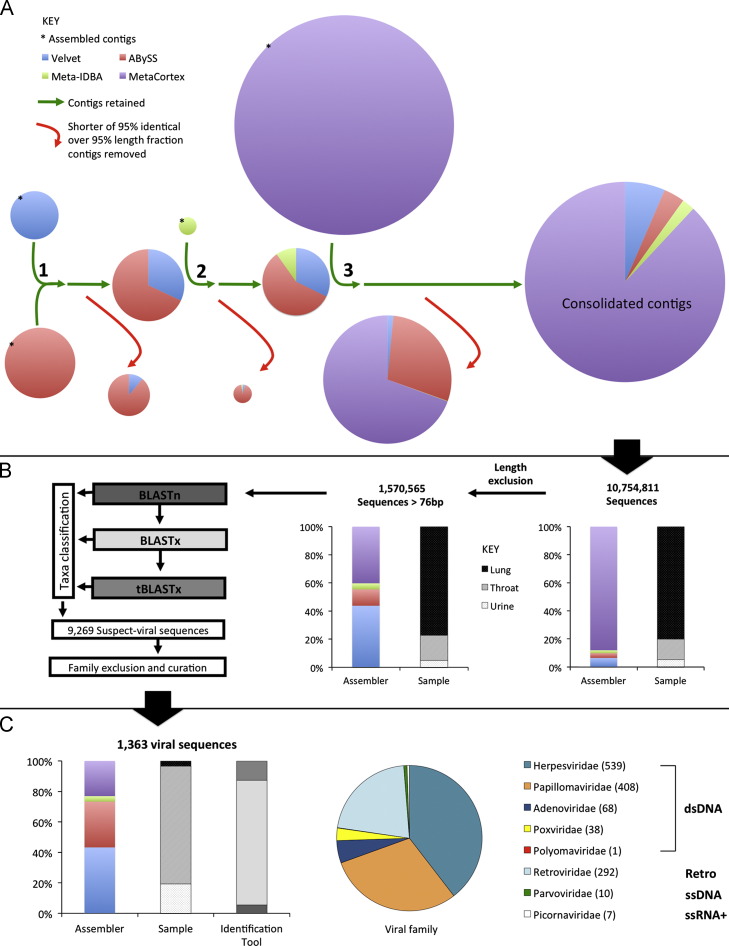
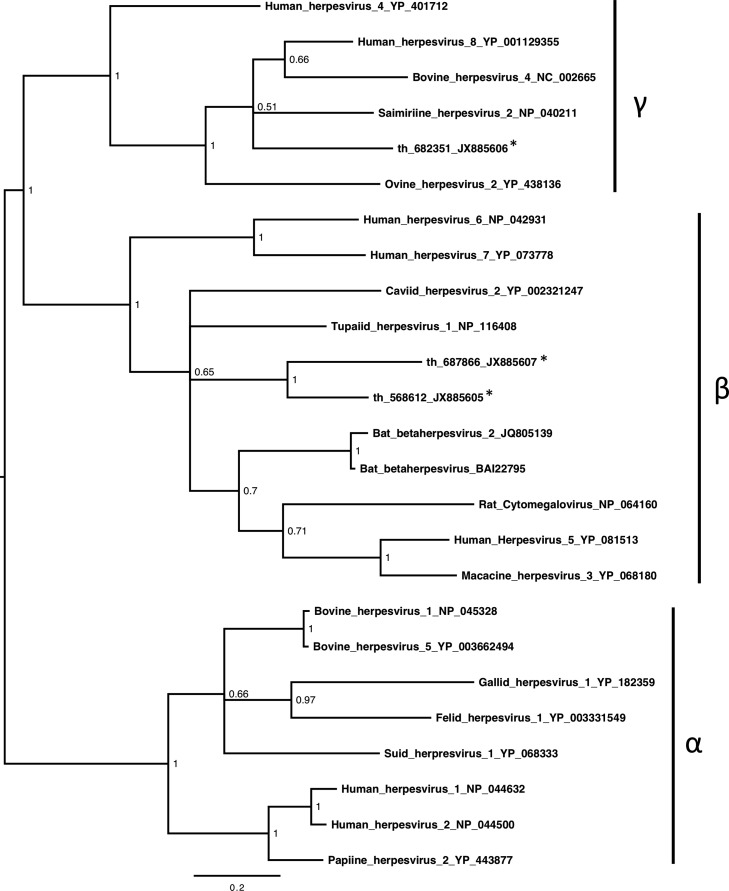
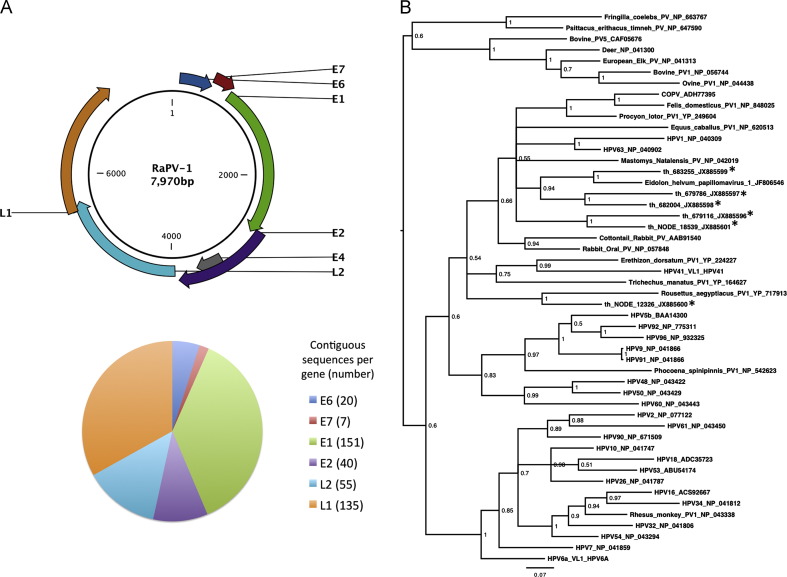
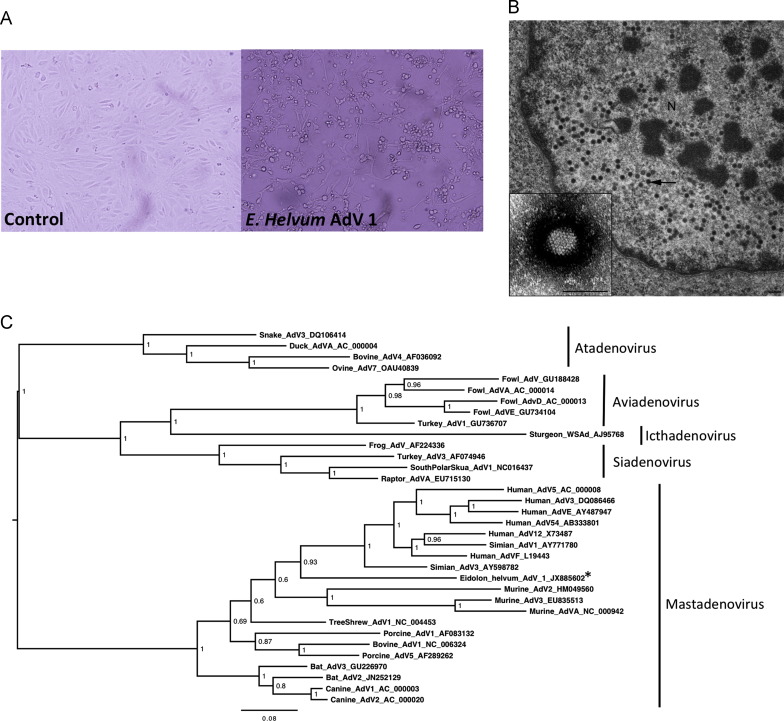
References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Bermingham, A., Chand, M.A., Brown, C.S., Aarons, E., Tong, C., Langrish, C., Hoschler, K., Brown, K., Galiano, M., Myers, R., Pebody, R.G., Green, H.J., Boddington, N.L., Gopal, R., Price, N., Newsholme, W., Drosten, C., Fouchier, R.A., Zambon, M., 2012. Severe Respiratory Illness Caused by a Novel Coronavirus, in a Patient Transferred to the United Kingdom from the Middle East, September 2012. Eurosurveillance 17, Available online: 〈http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20290〉. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
