

The fat side of prostate cancer
- PMID: 23562839
- PMCID: PMC3766375
- DOI: 10.1016/j.bbalip.2013.03.010
The fat side of prostate cancer
Abstract
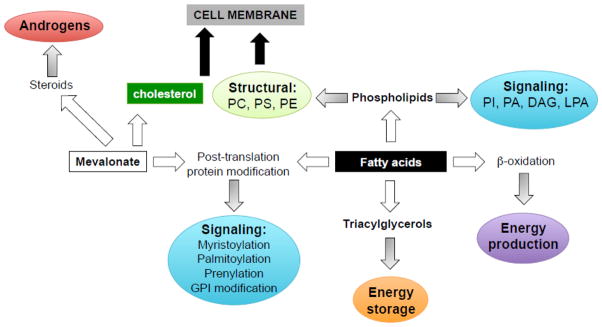
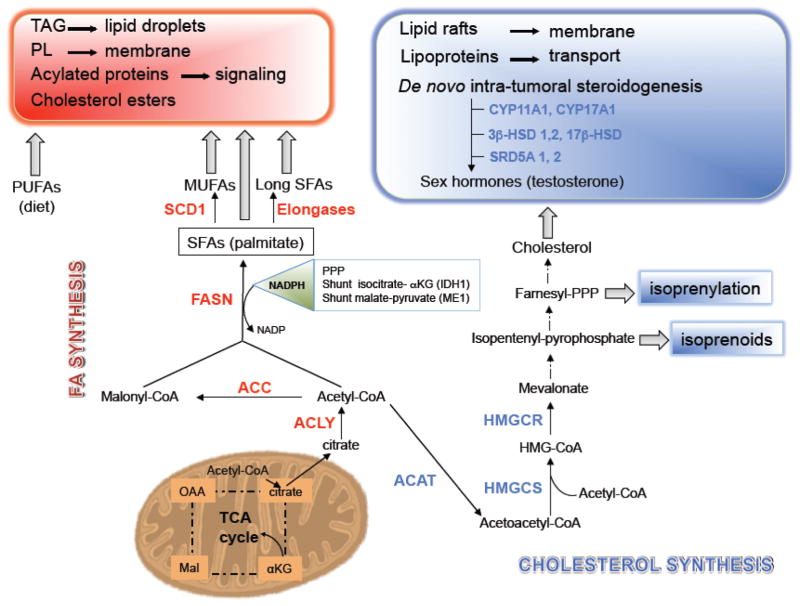
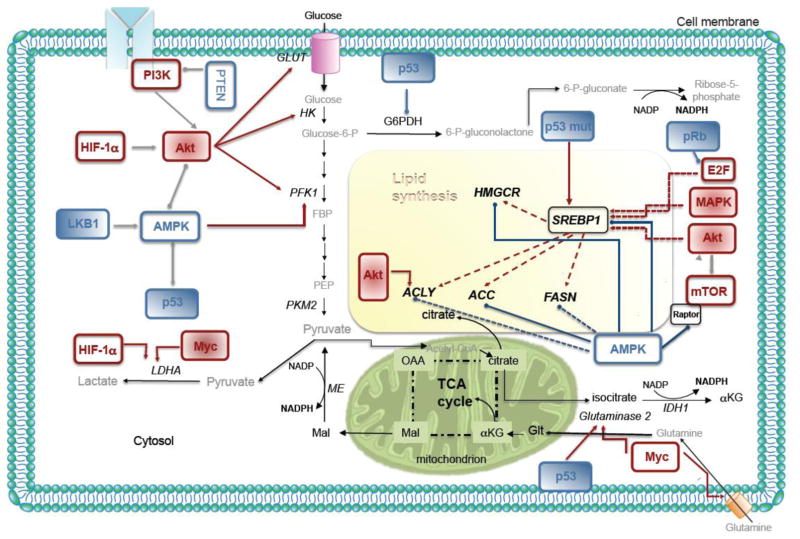
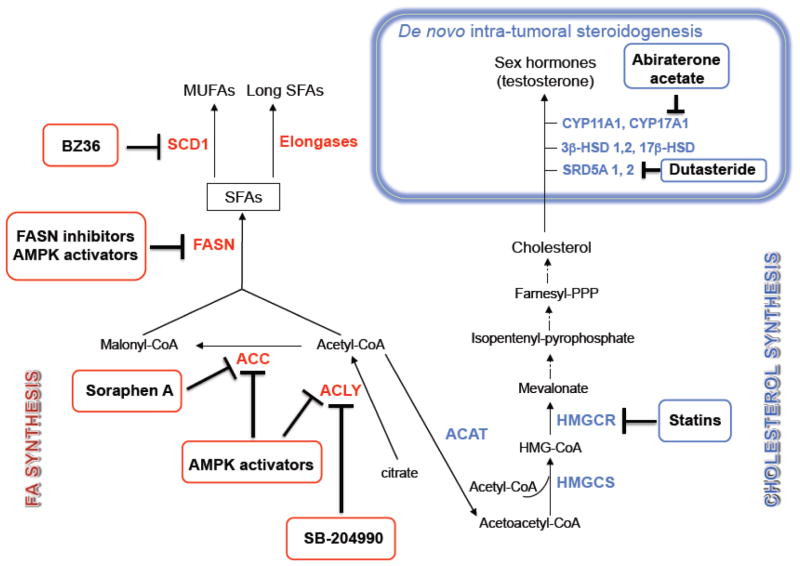
Prostate cancer (PCa) metabolism appears to be unique in comparison with other types of solid cancers. Normal prostate cells mainly rely on glucose oxidation to provide precursors for the synthesis and secretion of citrate, resulting in an incomplete Krebs cycle and minimal oxidative phosphorylation for energy production. In contrast, during transformation, PCa cells no longer secrete citrate and they reactivate the Krebs cycle as energy source. Moreover, primary PCas do not show increased aerobic glycolysis and therefore they are not efficiently detectable with (18)F-FDG-PET. However, increased de novo lipid synthesis, strictly intertwined with deregulation in classical oncogenes and oncosuppressors, is an early event of the disease. Up-regulation and increased activity of lipogenic enzymes (including fatty acid synthase and choline kinase) occurs throughout PCa carcinogenesis and correlates with worse prognosis and poor survival. Thus, lipid precursors such as acetate and choline have been successfully used as alternative tracers for PET imaging. Lipid synthesis intermediates and FA catabolism also emerged as important players in PCa maintenance. Finally, epidemiologic studies suggested that systemic metabolic disorders including obesity, metabolic syndrome, and diabetes as well as hypercaloric and fat-rich diets might increase the risk of PCa. However, how metabolic disorders contribute to PCa development and whether dietary lipids and de novo lipids synthesized intra-tumor are differentially metabolized still remains unclear. In this review, we examine the switch in lipid metabolism supporting the development and progression of PCa and we discuss how we can exploit its lipogenic nature for therapeutic and diagnostic purposes. This article is part of a Special Issue entitled Lipid Metabolism in Cancer.
Keywords: Fatty acids; Imaging; Lipid metabolism; Metabolic diseases; Prostate cancer.
Copyright © 2013 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
References
-
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61:212–236. - PubMed
-
- Djulbegovic M, Neuberger MM, Dahm P. Prostate-cancer mortality after PSA screening. The New England journal of medicine. 2012;366:2228–2229. author reply 2230–2221. - PubMed
-
- Bracarda S, Logothetis C, Sternberg CN, Oudard S. Current and emerging treatment modalities for metastatic castration-resistant prostate cancer. BJU international. 2011;107(Suppl 2):13–20. - PubMed
-
- Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. The New England journal of medicine. 2004;351:1513–1520. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
