

Smoothened is a master regulator of adult liver repair
- PMID: 23563311
- PMCID: PMC3668809
- DOI: 10.1172/JCI66904
Smoothened is a master regulator of adult liver repair
Abstract
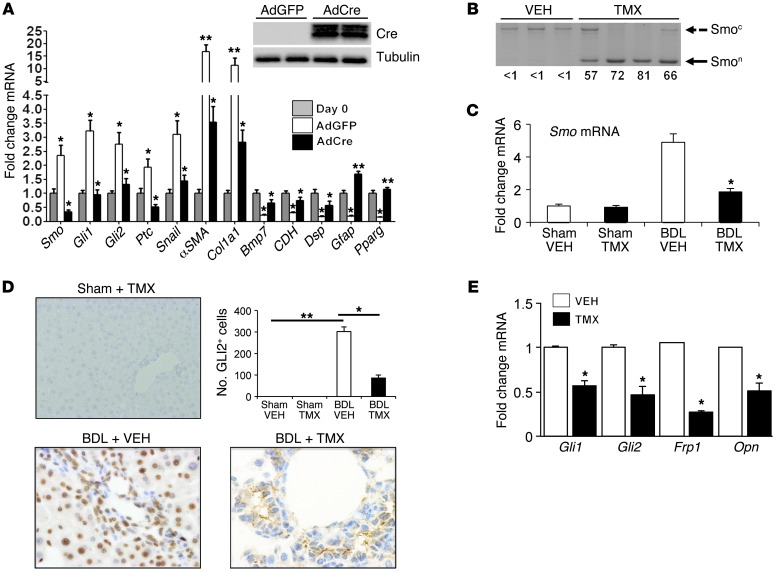
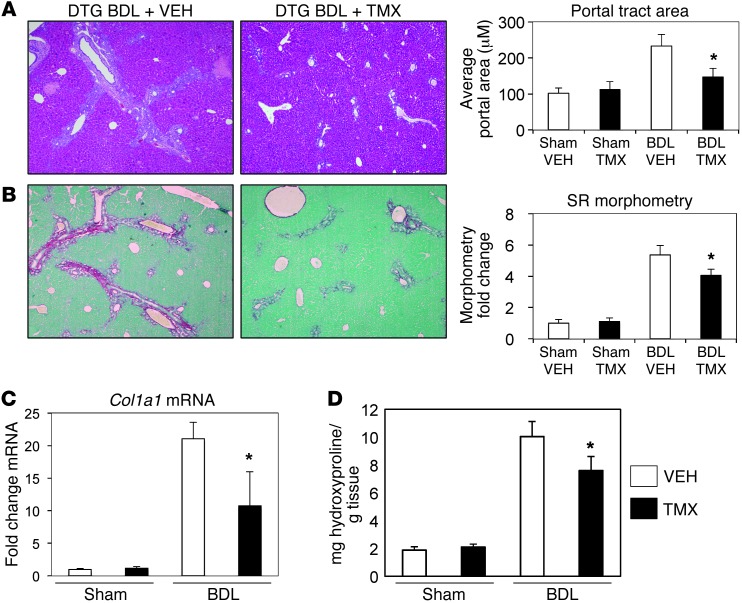
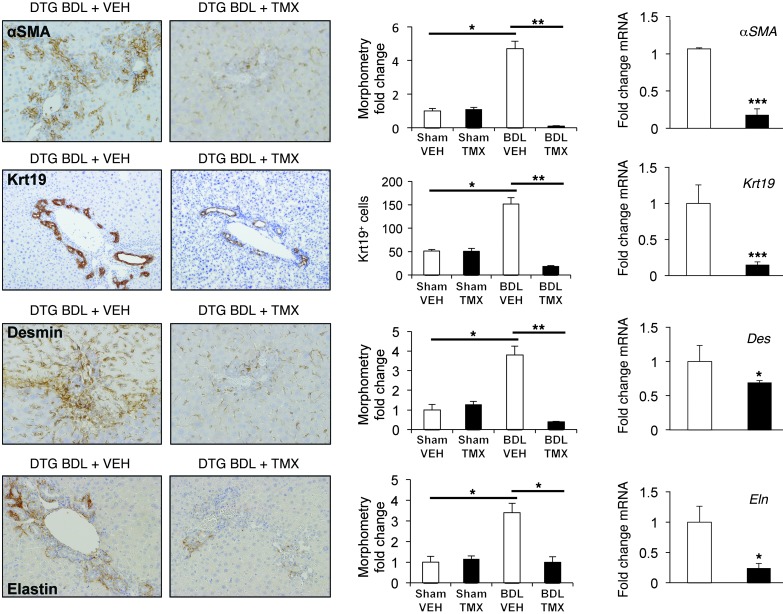
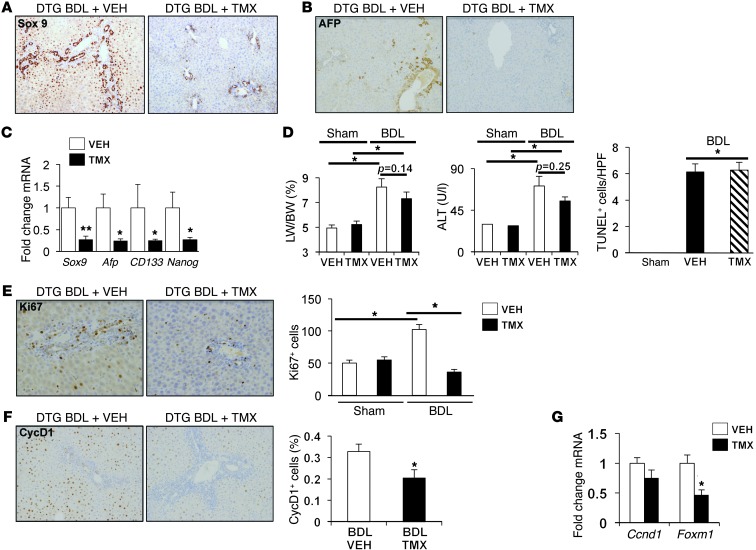
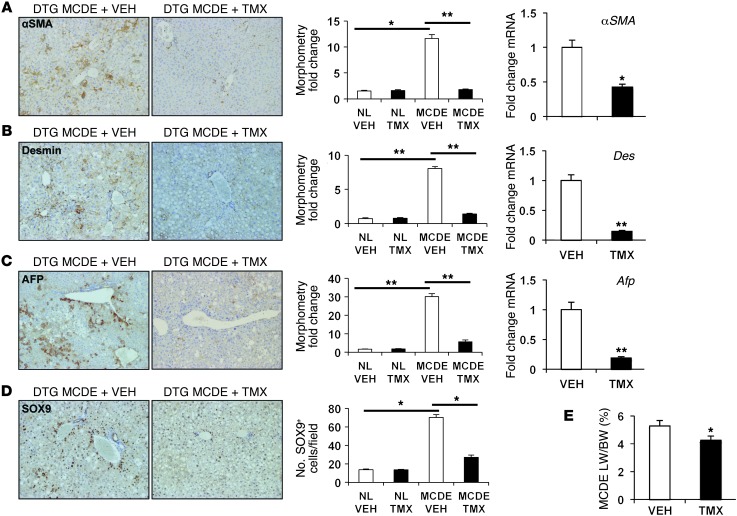
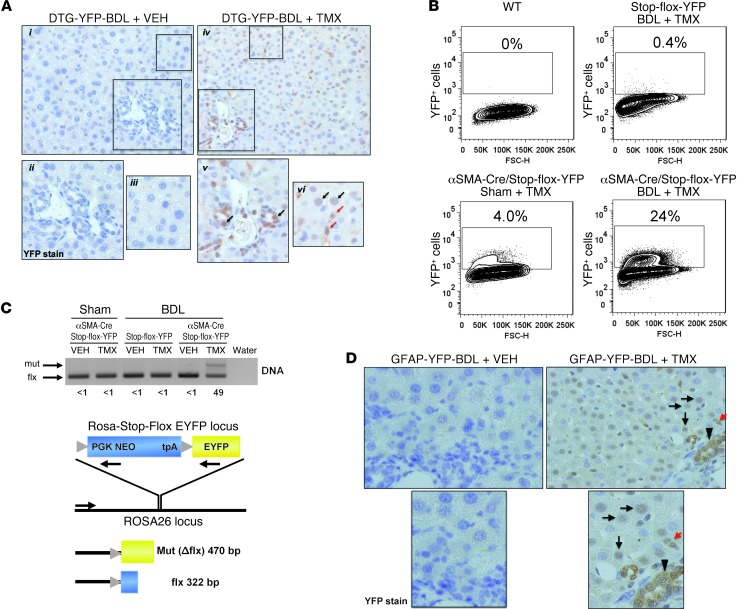
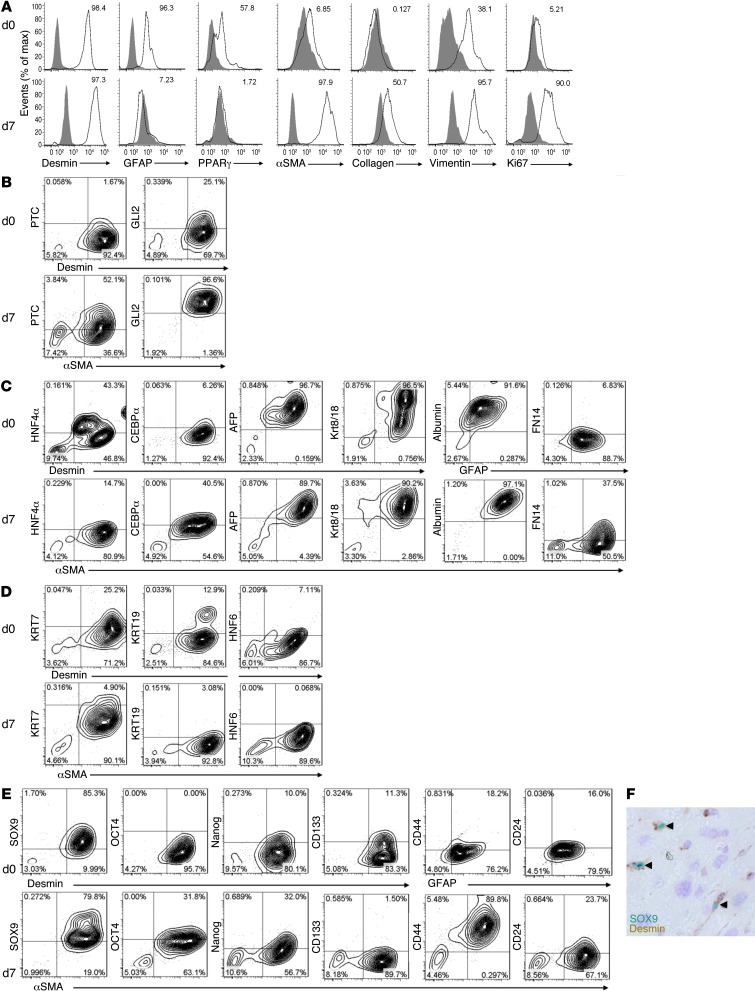
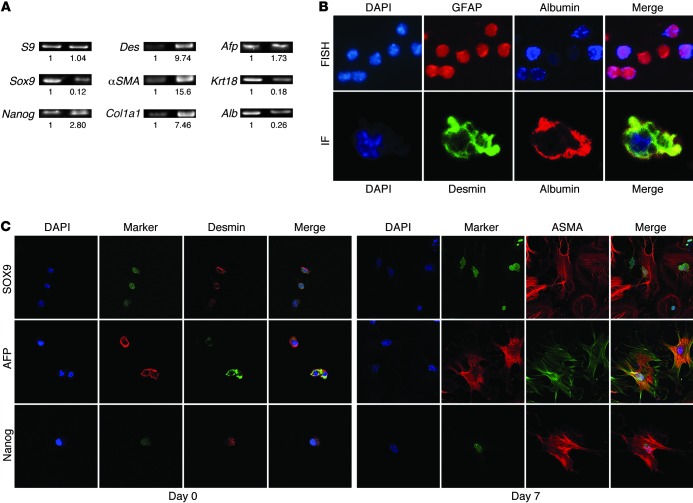
When regenerative processes cannot keep pace with cell death, functional epithelia are replaced by scar. Scarring is characterized by both excessive accumulation of fibrous matrix and persistent outgrowth of cell types that accumulate transiently during successful wound healing, including myofibroblasts (MFs) and progenitors. This suggests that signaling that normally directs these cells to repair injured epithelia is deregulated. To evaluate this possibility, we examined liver repair during different types of liver injury after Smoothened (SMO), an obligate intermediate in the Hedgehog (Hh) signaling pathway, was conditionally deleted in cells expressing the MF-associated gene, αSMA. Surprisingly, blocking canonical Hh signaling in MFs not only inhibited liver fibrosis but also prevented accumulation of liver progenitors. Hh-sensitive, hepatic stellate cells (HSCs) were identified as the source of both MFs and progenitors by lineage-tracing studies in 3 other strains of mice, coupled with analysis of highly pure HSC preparations using flow cytometry, immunofluorescence confocal microscopy, RT-PCR, and in situ hybridization. The results identify SMO as a master regulator of hepatic epithelial regeneration based on its ability to promote mesenchymal-to-epithelial transitions in a subpopulation of HSC-derived MFs with features of multipotent progenitors.
Figures
Similar articles
-
Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice.Hepatology. 2013 Nov;58(5):1801-13. doi: 10.1002/hep.26511. Epub 2013 Sep 30. Hepatology. 2013. PMID: 23703657 Free PMC article.
-
Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy.Gut. 2014 Aug;63(8):1333-44. doi: 10.1136/gutjnl-2013-305962. Epub 2013 Oct 30. Gut. 2014. PMID: 24173292 Free PMC article.
-
The intrahepatic signalling niche of hedgehog is defined by primary cilia positive cells during chronic liver injury.J Hepatol. 2014 Jan;60(1):143-51. doi: 10.1016/j.jhep.2013.08.012. Epub 2013 Aug 23. J Hepatol. 2014. PMID: 23978713
-
Wnt signaling in liver fibrosis: progress, challenges and potential directions.Biochimie. 2013 Dec;95(12):2326-35. doi: 10.1016/j.biochi.2013.09.003. Epub 2013 Sep 13. Biochimie. 2013. PMID: 24036368 Review.
-
MicroRNAs in liver fibrosis: Focusing on the interaction with hedgehog signaling.World J Gastroenterol. 2016 Aug 7;22(29):6652-62. doi: 10.3748/wjg.v22.i29.6652. World J Gastroenterol. 2016. PMID: 27547008 Free PMC article. Review.
Cited by
-
The hedgehog pathway suppresses neuropathogenesis in CD4 T cell-driven inflammation.Brain. 2021 Jul 28;144(6):1670-1683. doi: 10.1093/brain/awab083. Brain. 2021. PMID: 33723591 Free PMC article.
-
Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice.Hepatology. 2013 Nov;58(5):1801-13. doi: 10.1002/hep.26511. Epub 2013 Sep 30. Hepatology. 2013. PMID: 23703657 Free PMC article.
-
Transcriptional regulation of Hepatic Stellate Cell activation in NASH.Sci Rep. 2019 Feb 20;9(1):2324. doi: 10.1038/s41598-019-39112-6. Sci Rep. 2019. PMID: 30787418 Free PMC article.
-
Evidence for and against epithelial-to-mesenchymal transition in the liver.Am J Physiol Gastrointest Liver Physiol. 2013 Dec;305(12):G881-90. doi: 10.1152/ajpgi.00289.2013. Epub 2013 Oct 24. Am J Physiol Gastrointest Liver Physiol. 2013. PMID: 24157970 Free PMC article.
-
Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte.Liver Int. 2015 Feb;35(2):302-10. doi: 10.1111/liv.12577. Epub 2014 May 20. Liver Int. 2015. PMID: 24766136 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
