

Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci
- PMID: 23570689
- PMCID: PMC3698506
- DOI: 10.1101/gr.155127.113
Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci
Abstract
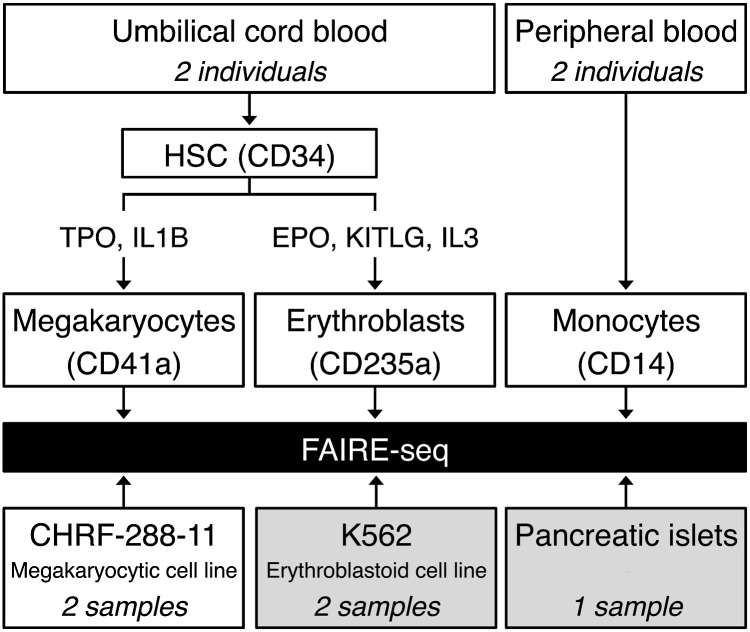
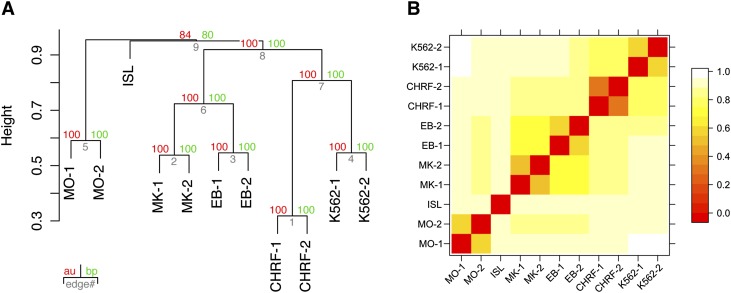
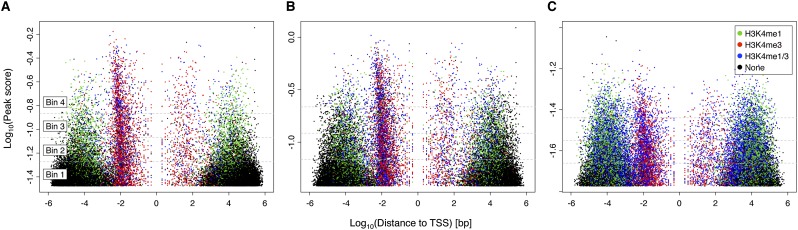
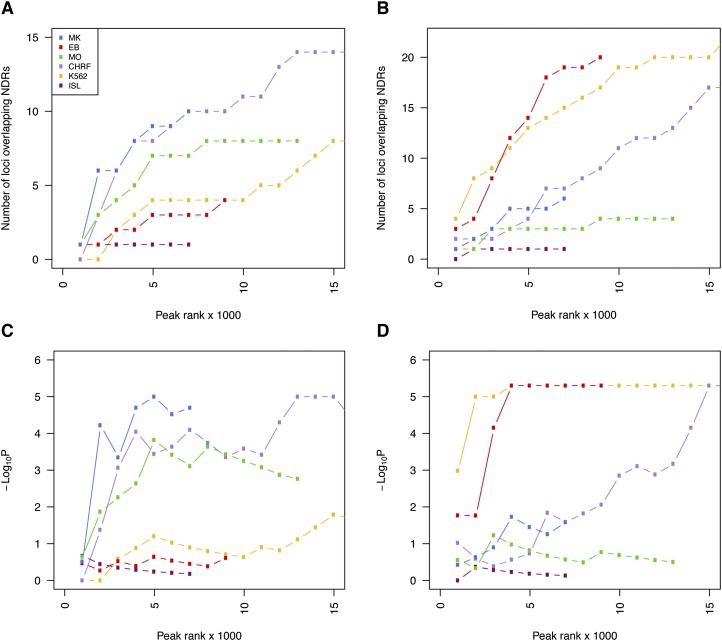
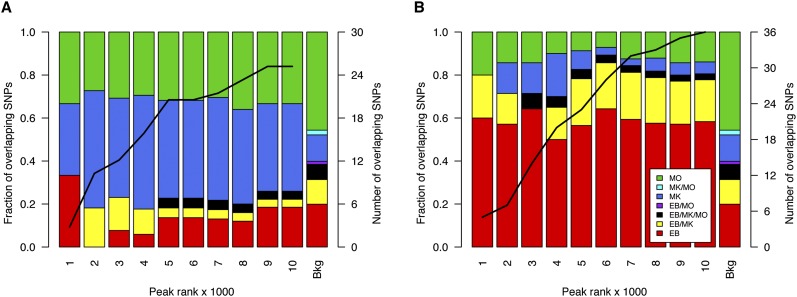
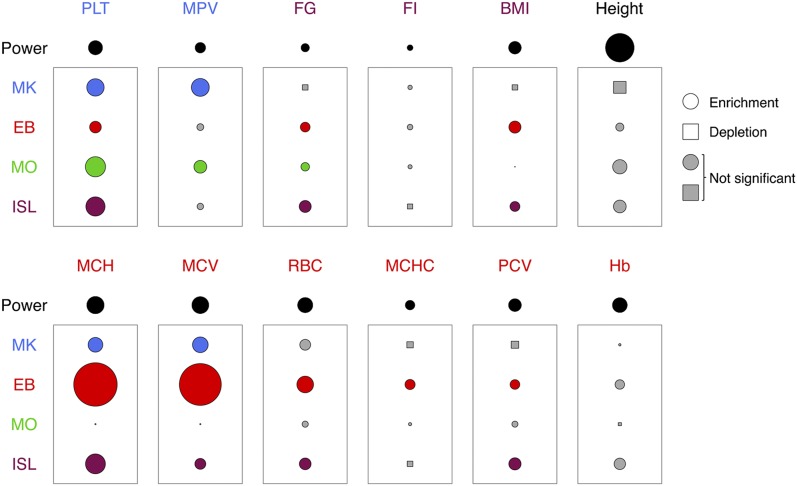
Nearly three-quarters of the 143 genetic signals associated with platelet and erythrocyte phenotypes identified by meta-analyses of genome-wide association (GWA) studies are located at non-protein-coding regions. Here, we assessed the role of candidate regulatory variants associated with cell type-restricted, closely related hematological quantitative traits in biologically relevant hematopoietic cell types. We used formaldehyde-assisted isolation of regulatory elements followed by next-generation sequencing (FAIRE-seq) to map regions of open chromatin in three primary human blood cells of the myeloid lineage. In the precursors of platelets and erythrocytes, as well as in monocytes, we found that open chromatin signatures reflect the corresponding hematopoietic lineages of the studied cell types and associate with the cell type-specific gene expression patterns. Dependent on their signal strength, open chromatin regions showed correlation with promoter and enhancer histone marks, distance to the transcription start site, and ontology classes of nearby genes. Cell type-restricted regions of open chromatin were enriched in sequence variants associated with hematological indices. The majority (63.6%) of such candidate functional variants at platelet quantitative trait loci (QTLs) coincided with binding sites of five transcription factors key in regulating megakaryopoiesis. We experimentally tested 13 candidate regulatory variants at 10 platelet QTLs and found that 10 (76.9%) affected protein binding, suggesting that this is a frequent mechanism by which regulatory variants influence quantitative trait levels. Our findings demonstrate that combining large-scale GWA data with open chromatin profiles of relevant cell types can be a powerful means of dissecting the genetic architecture of closely related quantitative traits.
Figures
References
-
- Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, Bock C, Boehm B, Campo E, Caricasole A, et al. 2012. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol 30: 224–226 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases