

Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications
- PMID: 23571099
- PMCID: PMC3654056
- DOI: 10.1016/j.taap.2013.03.026
Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications
Abstract
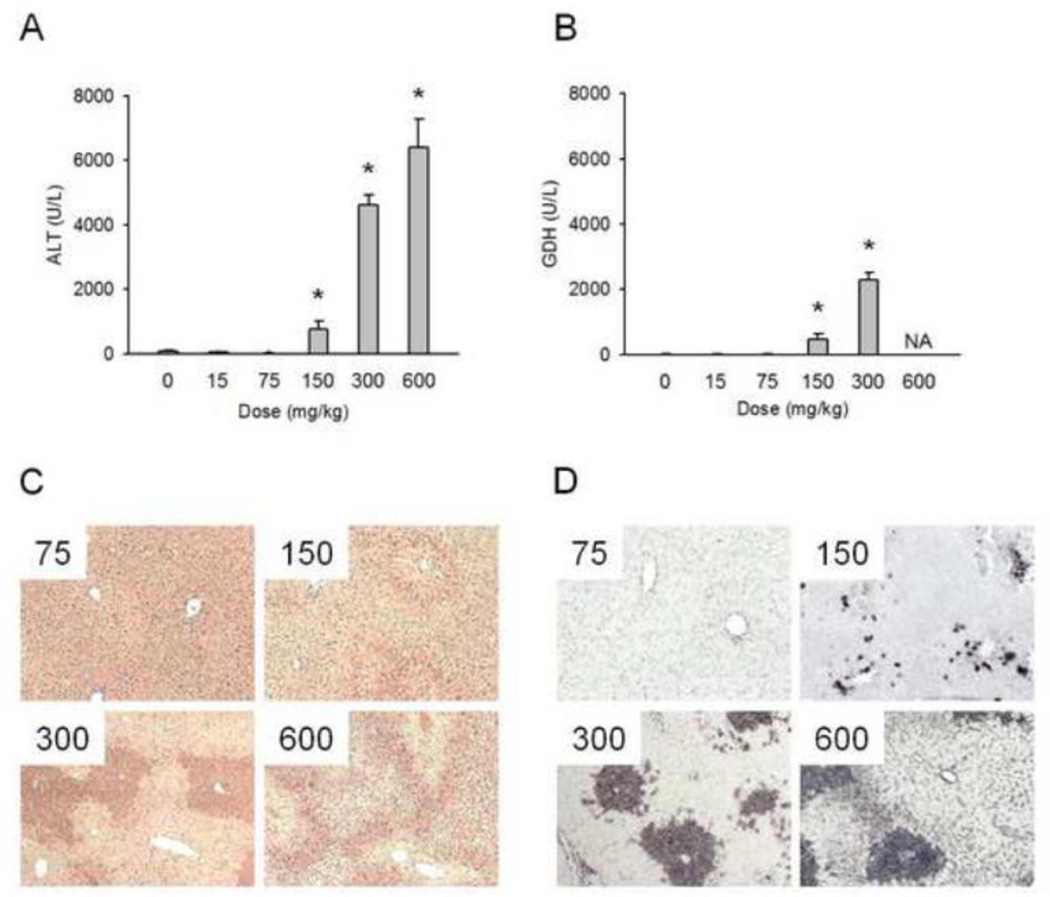
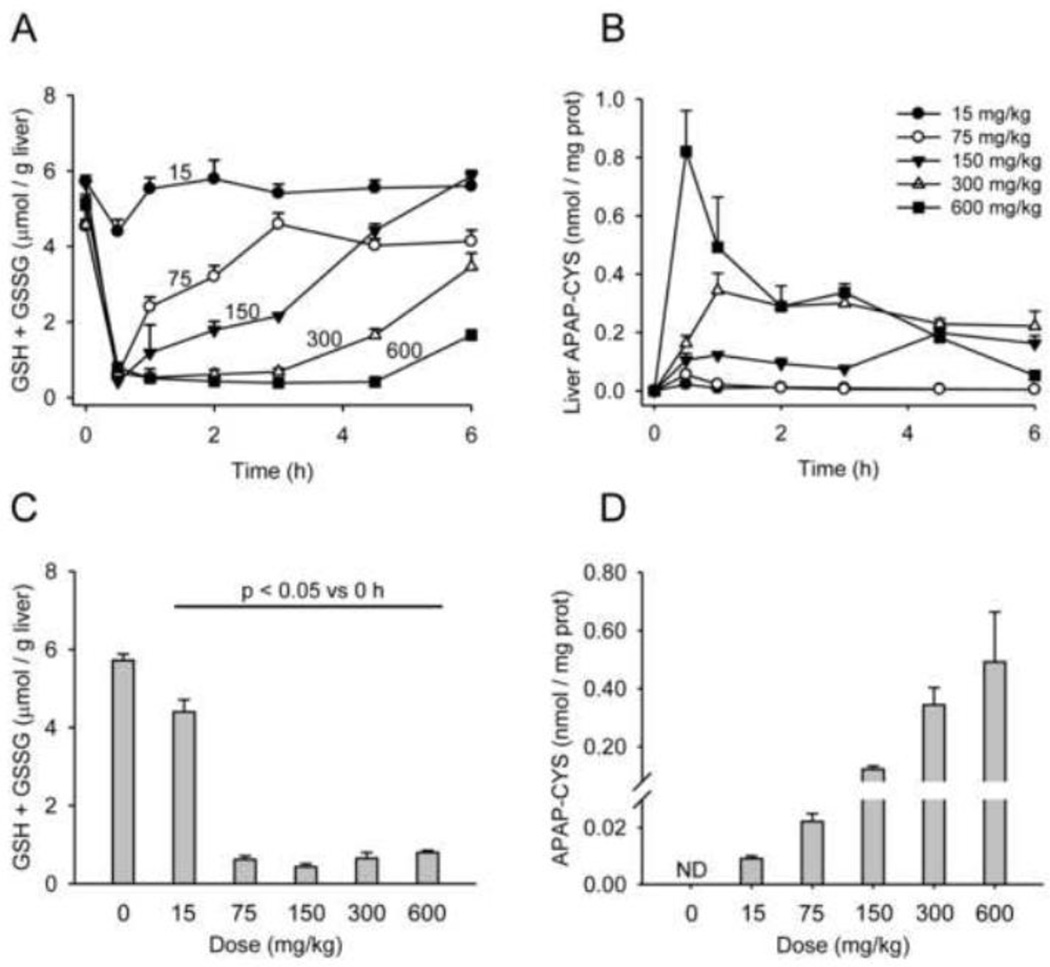
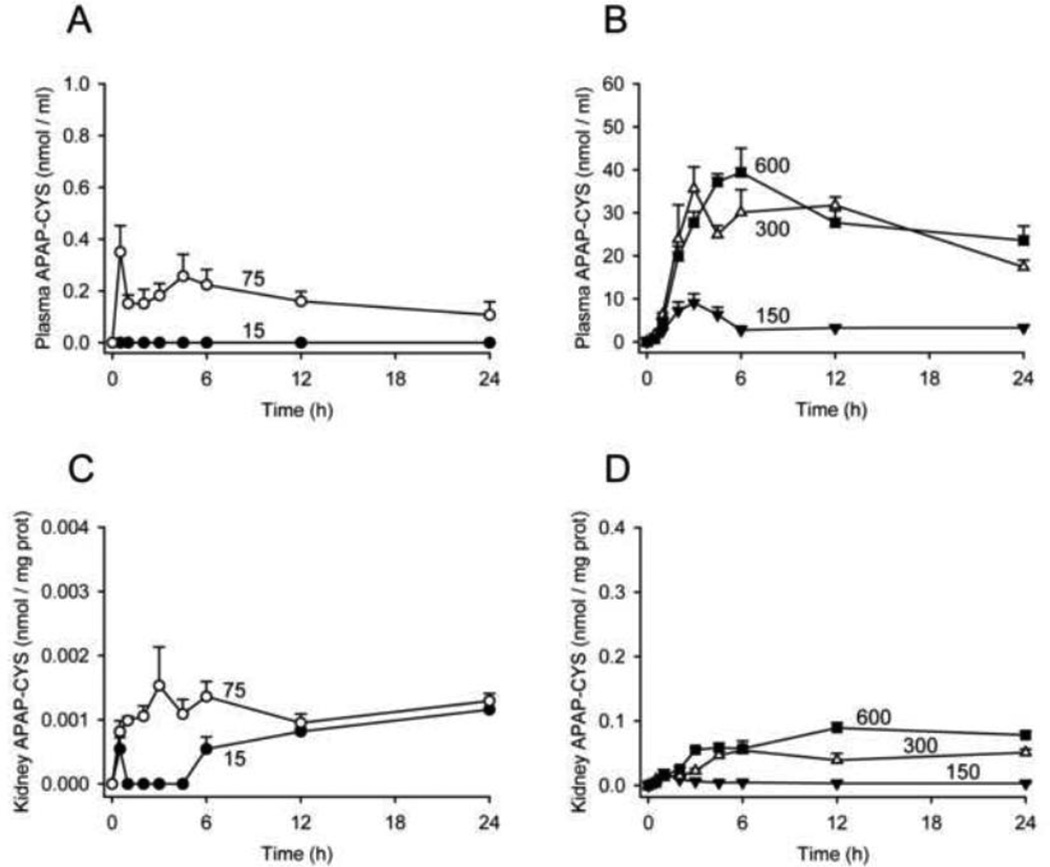
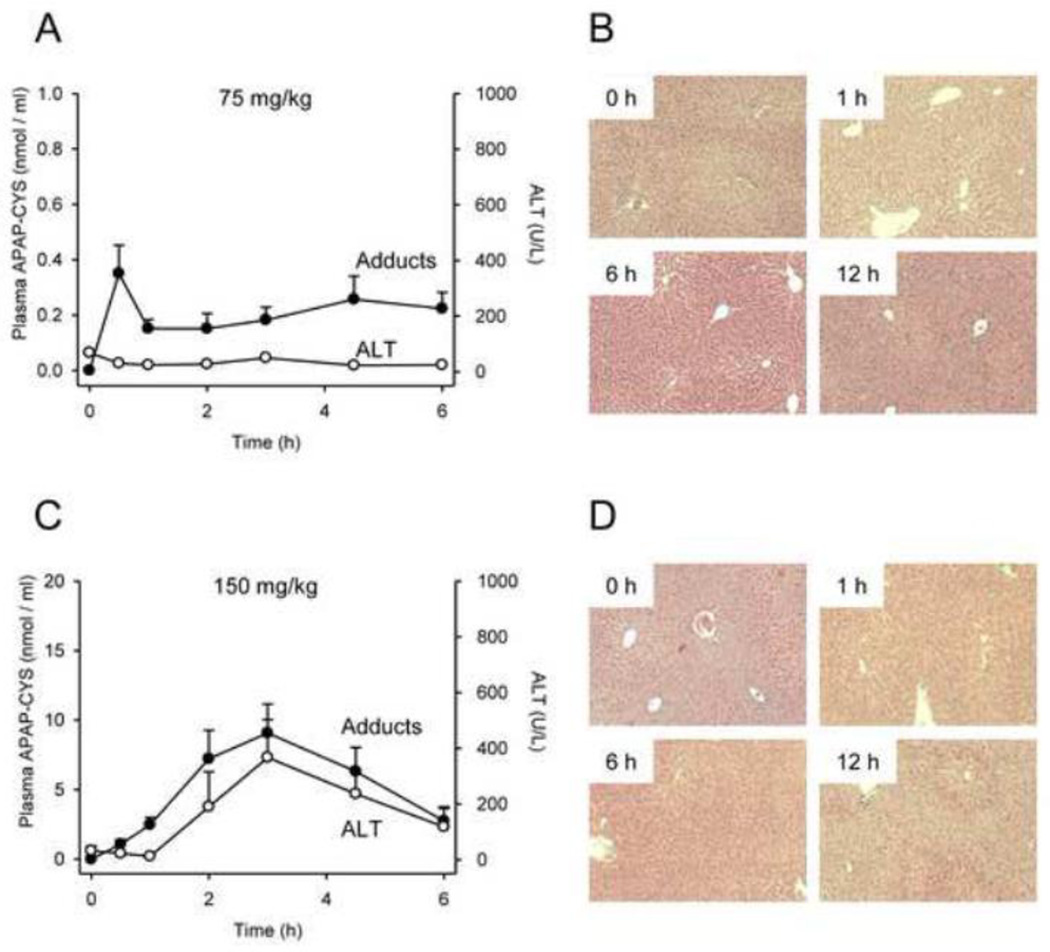
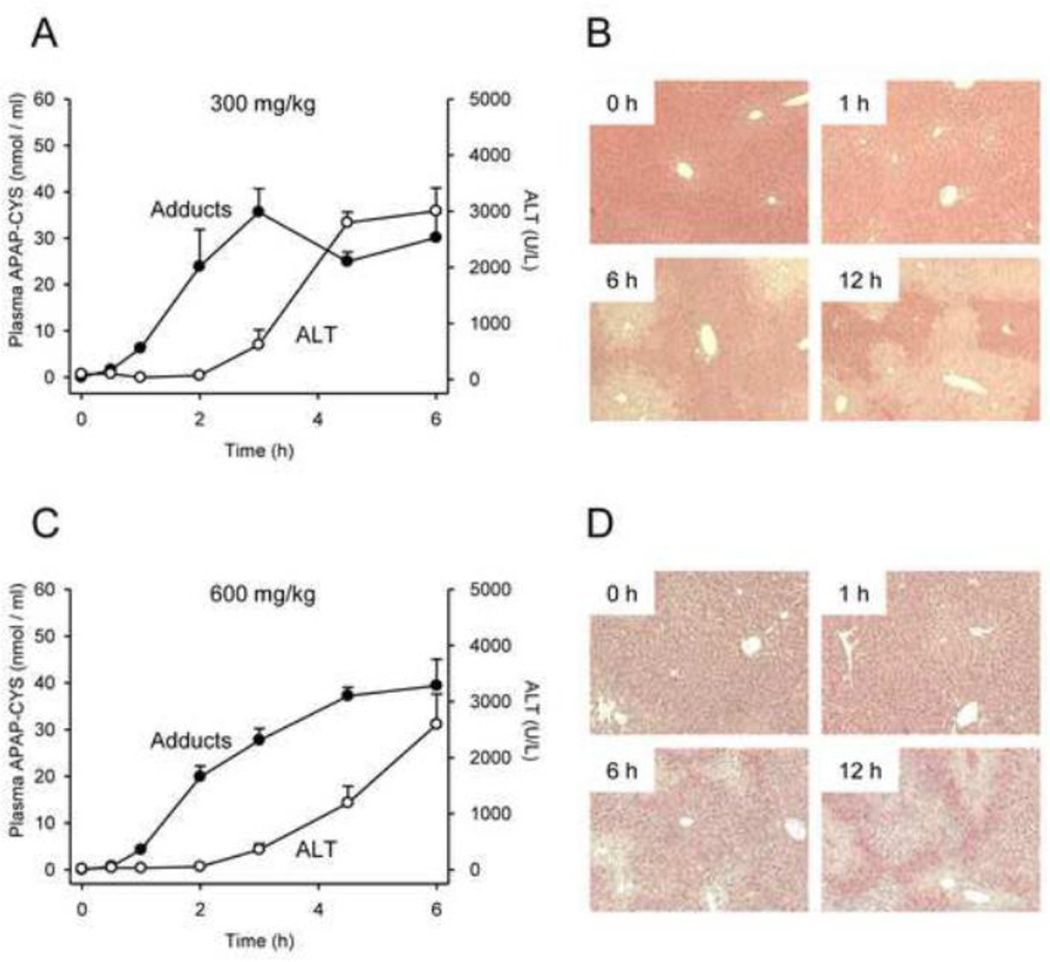
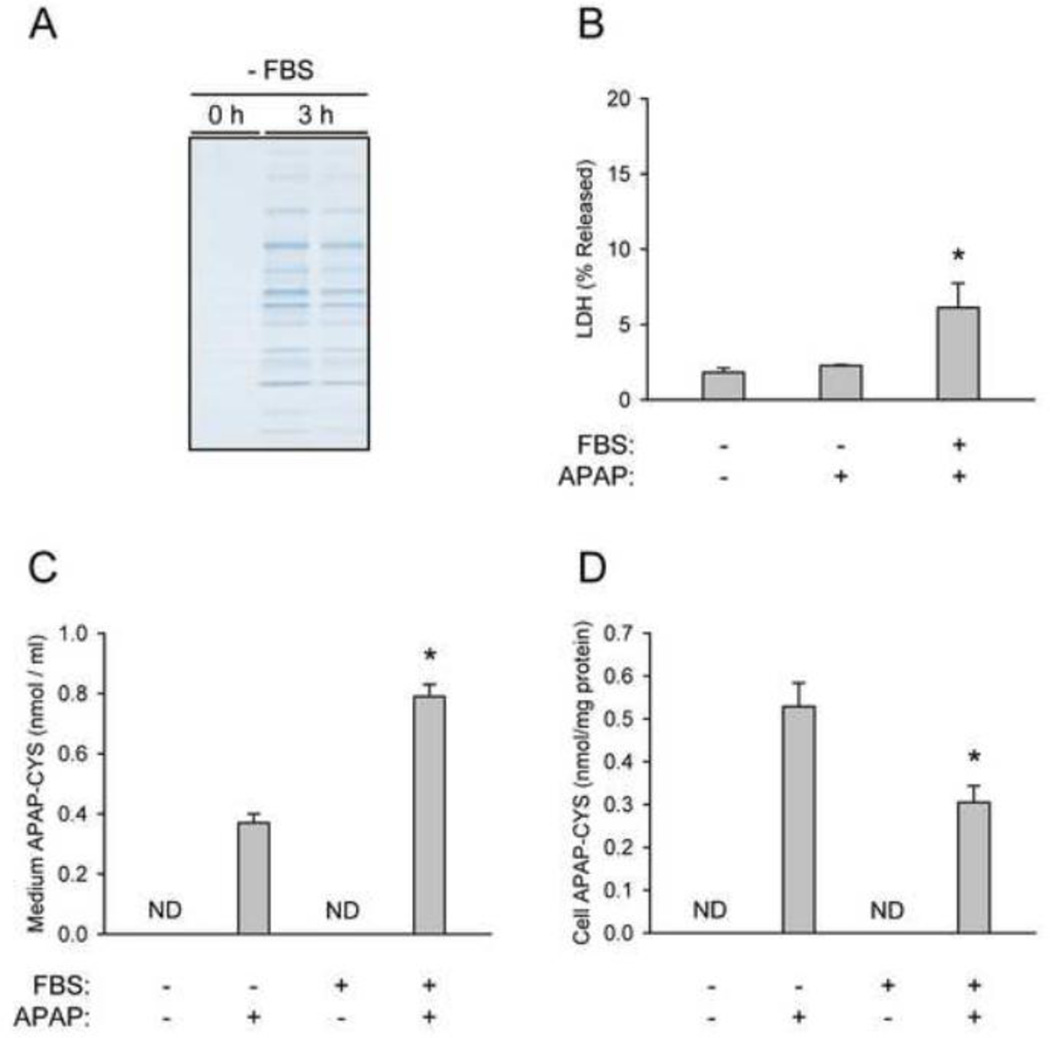
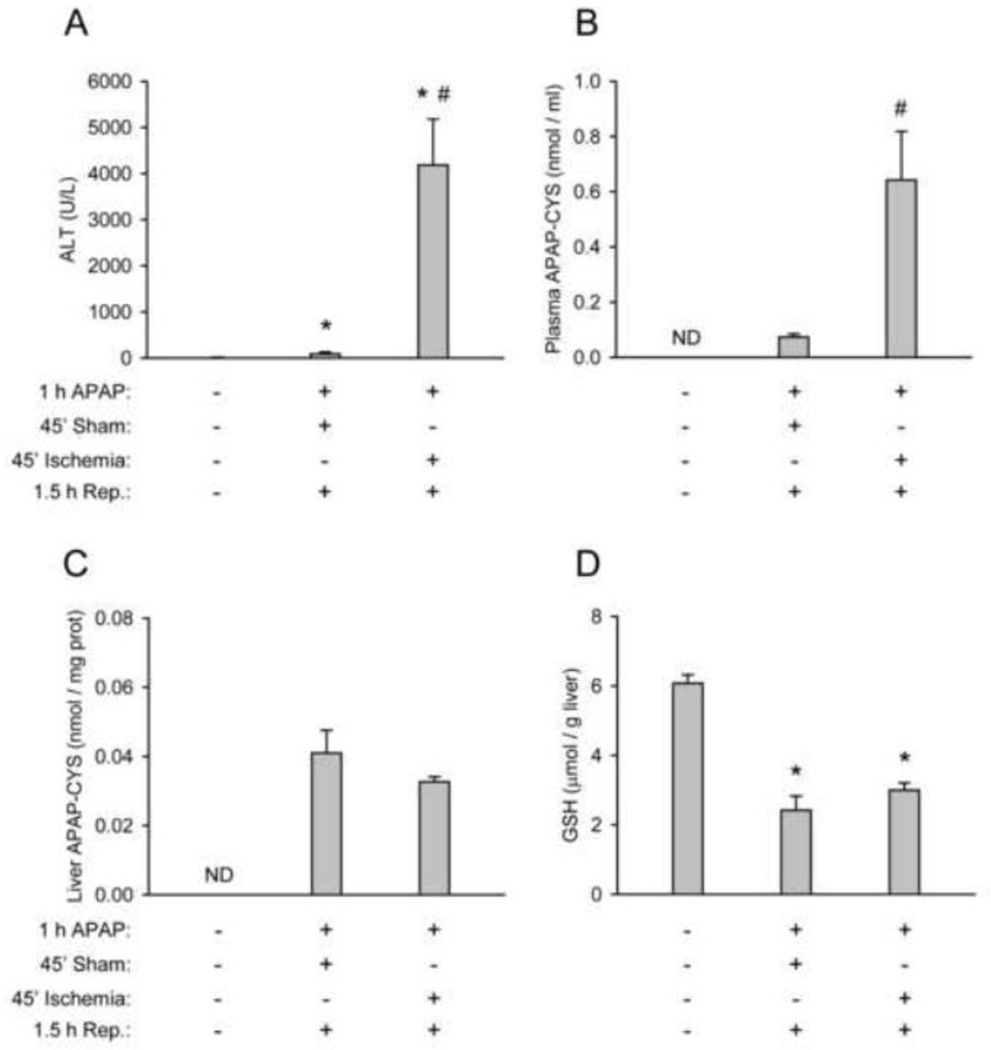
At therapeutic doses, acetaminophen (APAP) is a safe and effective analgesic. However, overdose of APAP is the principal cause of acute liver failure in the West. Binding of the reactive metabolite of APAP (NAPQI) to proteins is thought to be the initiating event in the mechanism of hepatotoxicity. Early work suggested that APAP-protein binding could not occur without glutathione (GSH) depletion, and likely only at toxic doses. Moreover, it was found that protein-derived APAP-cysteine could only be detected in serum after the onset of liver injury. On this basis, it was recently proposed that serum APAP-cysteine could be used as diagnostic marker of APAP overdose. However, comprehensive dose-response and time course studies have not yet been done. Furthermore, the effects of co-morbidities on this parameter have not been investigated. We treated groups of mice with APAP at multiple doses and measured liver GSH and both liver and plasma APAP-protein adducts at various timepoints. Our results show that protein binding can occur without much loss of GSH. Importantly, the data confirm earlier work that showed that protein-derived APAP-cysteine can appear in plasma without liver injury. Experiments performed in vitro suggest that this may involve multiple mechanisms, including secretion of adducted proteins and diffusion of NAPQI directly into plasma. Induction of liver necrosis through ischemia-reperfusion significantly increased the plasma concentration of protein-derived APAP-cysteine after a subtoxic dose of APAP. While our data generally support the measurement of serum APAP-protein adducts in the clinic, caution is suggested in the interpretation of this parameter.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK. Molecular forms of HMGB1 and keratin-8 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. patol. 2012;56:1070–1079. - PMC - PubMed
-
- Axworthy DB, Hoffmann KJ, Streeter AJ, Calleman CJ, Pascoe GA, Baillie TA. Covalent binding of acetaminophen to mouse hemoglobin. Identification of major and minor adducts formed in vivo and implications for the nature of the arylating metabolites. Chem. Biol. Interact. 1988;68:99–116. - PubMed
-
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol. Sci. 2006;94:217–225. - PubMed
-
- Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2008;324:8–14. - PubMed
-
- Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol. Sci. 2004;80:343–349. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
