

Long QT syndrome-associated mutations in intrauterine fetal death
- PMID: 23571586
- PMCID: PMC3852902
- DOI: 10.1001/jama.2013.3219
Long QT syndrome-associated mutations in intrauterine fetal death
Abstract
Importance: Intrauterine fetal death or stillbirth occurs in approximately 1 out of every 160 pregnancies and accounts for 50% of all perinatal deaths. Postmortem evaluation fails to elucidate an underlying cause in many cases. Long QT syndrome (LQTS) may contribute to this problem.
Objective: To determine the spectrum and prevalence of mutations in the 3 most common LQTS susceptible genes (KCNQ1, KCNH2, and SCN5A) for a cohort of unexplained cases.
Design, setting, and patients: In this case series, retrospective postmortem genetic testing was conducted on a convenience sample of 91 unexplained intrauterine fetal deaths (mean [SD] estimated gestational age at fetal death, 26.3 [8.7] weeks) that were collected from 2006-2012 by the Mayo Clinic, Rochester, Minnesota, or the Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. More than 1300 ostensibly healthy individuals served as controls. In addition, publicly available exome databases were assessed for the general population frequency of identified genetic variants.
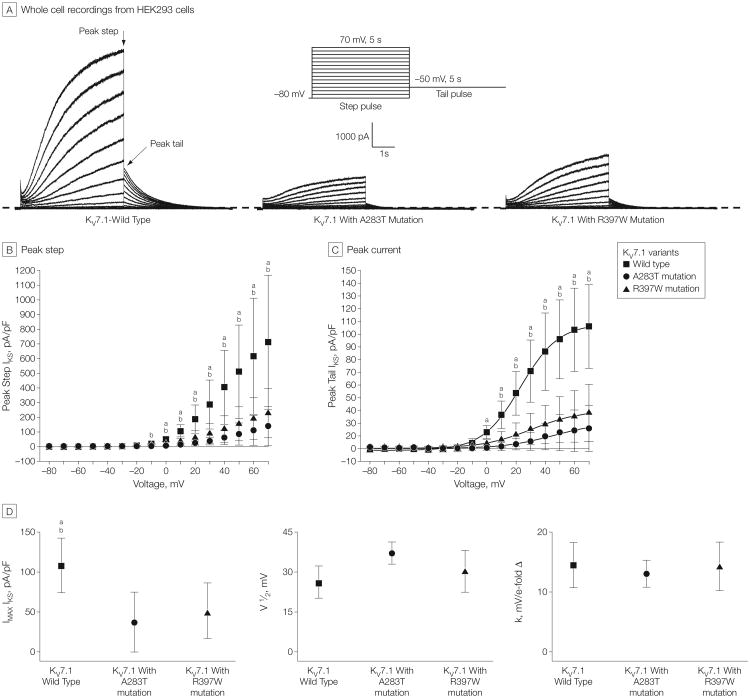
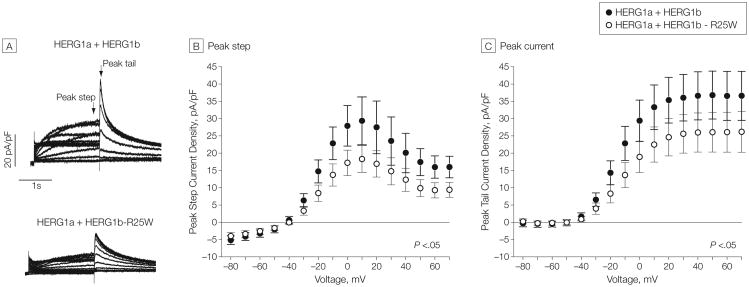
Main outcomes and measures: Comprehensive mutational analyses of KCNQ1 (KV7.1, LQTS type 1), KCNH2 (HERG/KV11.1, LQTS type 2), and SCN5A (NaV1.5, LQTS type 3) were performed using denaturing high-performance liquid chromatography and direct DNA sequencing on genomic DNA extracted from decedent tissue. Functional analyses of novel mutations were performed using heterologous expression and patch-clamp recording.
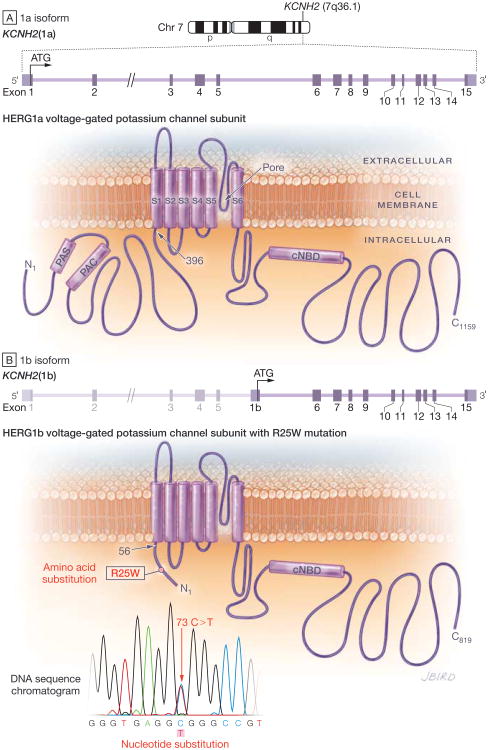
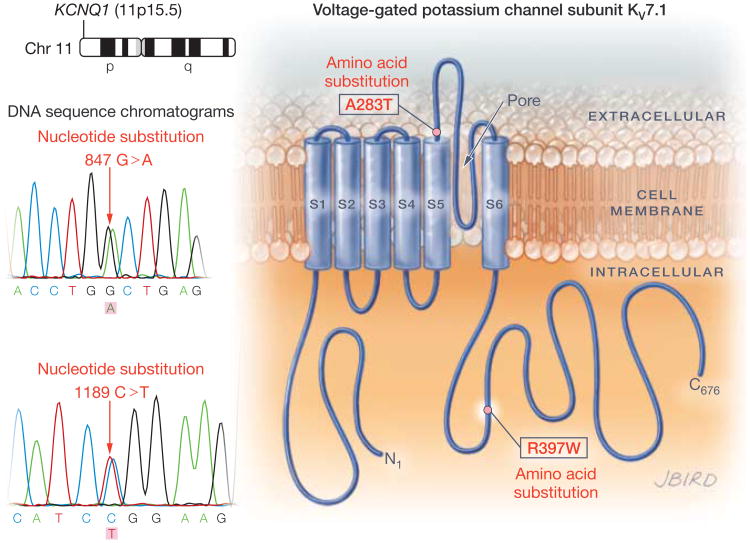
Results: The 3 putative LQTS susceptibility missense mutations (KCNQ1, p.A283T; KCNQ1, p.R397W; and KCNH2 [1b], p.R25W), with a heterozygous frequency of less than 0.05% in more than 10 000 publicly available exomes and absent in more than 1000 ethnically similar control patients, were discovered in 3 intrauterine fetal deaths (3.3% [95% CI, 0.68%-9.3%]). Both KV7.1-A283T (16-week male) and KV7.1-R397W (16-week female) mutations were associated with marked KV7.1 loss-of-function consistent with in utero LQTS type 1, whereas the HERG1b-R25W mutation (33.2-week male) exhibited a loss of function consistent with in utero LQTS type 2. In addition, 5 intrauterine fetal deaths hosted SCN5A rare nonsynonymous genetic variants (p.T220I, p.R1193Q, involving 2 cases, and p.P2006A, involving 2 cases) that conferred in vitro electrophysiological characteristics consistent with potentially proarrhythmic phenotypes.
Conclusions and relevance: In this molecular genetic evaluation of 91 cases of intrauterine fetal death, missense mutations associated with LQTS susceptibility were discovered in 3 cases (3.3%) and overall, genetic variants leading to dysfunctional LQTS-associated ion channels in vitro were discovered in 8 cases (8.8%). These preliminary findings may provide insights into mechanisms of some cases of stillbirth.
Conflict of interest statement
Figures
Comment in
-
Long QT syndrome susceptibility mutations and pregnancy loss: another piece of a still unfinished puzzle?JAMA. 2013 Apr 10;309(14):1525-6. doi: 10.1001/jama.2013.3373. JAMA. 2013. PMID: 23571592 No abstract available.
Similar articles
-
The prevalence of mutations in KCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases.J Cardiovasc Electrophysiol. 2012 Oct;23(10):1092-8. doi: 10.1111/j.1540-8167.2012.02371.x. Epub 2012 Aug 6. J Cardiovasc Electrophysiol. 2012. PMID: 22882672
-
Mutation Analysis of KCNQ1, KCNH2 and SCN5A Genes in Taiwanese Long QT Syndrome Patients.Int Heart J. 2015;56(4):450-3. doi: 10.1536/ihj.14-428. Epub 2015 Jun 26. Int Heart J. 2015. PMID: 26118593
-
Long QT and Brugada syndrome gene mutations in New Zealand.Heart Rhythm. 2007 Oct;4(10):1306-14. doi: 10.1016/j.hrthm.2007.06.022. Epub 2007 Jul 14. Heart Rhythm. 2007. PMID: 17905336
-
Genetics of long QT syndrome.Methodist Debakey Cardiovasc J. 2014 Jan-Mar;10(1):29-33. doi: 10.14797/mdcj-10-1-29. Methodist Debakey Cardiovasc J. 2014. PMID: 24932360 Free PMC article. Review.
-
Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes.Int J Mol Sci. 2022 Jul 2;23(13):7389. doi: 10.3390/ijms23137389. Int J Mol Sci. 2022. PMID: 35806392 Free PMC article. Review.
Cited by
-
In utero Diagnosis of Long QT Syndrome: Challenges, Progress, and the Future.J Pregnancy Child Health. 2015;3(1):e125. doi: 10.4172/2376-127X.1000e125. Epub 2015 Dec 20. J Pregnancy Child Health. 2015. PMID: 27689137 Free PMC article. No abstract available.
-
Recurrent Pregnancy Loss and Concealed Long-QT Syndrome.J Am Heart Assoc. 2021 Sep 7;10(17):e021236. doi: 10.1161/JAHA.121.021236. Epub 2021 Aug 16. J Am Heart Assoc. 2021. PMID: 34398675 Free PMC article.
-
Asymmetry of parental origin in long QT syndrome: preferential maternal transmission of KCNQ1 variants linked to channel dysfunction.Eur J Hum Genet. 2016 Aug;24(8):1160-6. doi: 10.1038/ejhg.2015.257. Epub 2015 Dec 16. Eur J Hum Genet. 2016. PMID: 26669661 Free PMC article.
-
Infection: the neglected paradigm in SIDS research.Arch Dis Child. 2017 Aug;102(8):767-772. doi: 10.1136/archdischild-2016-312327. Epub 2017 Jan 23. Arch Dis Child. 2017. PMID: 28115322 Free PMC article. Review.
-
Impact of genetics on the clinical management of channelopathies.J Am Coll Cardiol. 2013 Jul 16;62(3):169-180. doi: 10.1016/j.jacc.2013.04.044. Epub 2013 May 15. J Am Coll Cardiol. 2013. PMID: 23684683 Free PMC article. Review.
References
-
- Macdorman MF, Kirmeyer SE, Wilson EC. Fetal and Perinatal Mortality, United States, 2006. Natl Vital Stat Rep. 2012;60(8):1–22. - PubMed
-
- Cousens S, Blencowe H, Stanton C, et al. National, regional, and worldwide estimates of stillbirth rates in 2009 with trends since 1995: a systematic analysis. Lancet. 2011;377(9774):1319–1330. - PubMed
-
- Ackerman MJ, Siu BL, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286(18):2264–2269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
