

Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes
- PMID: 23575371
- PMCID: PMC3749498
- DOI: 10.1038/ismej.2013.59
Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes
Abstract
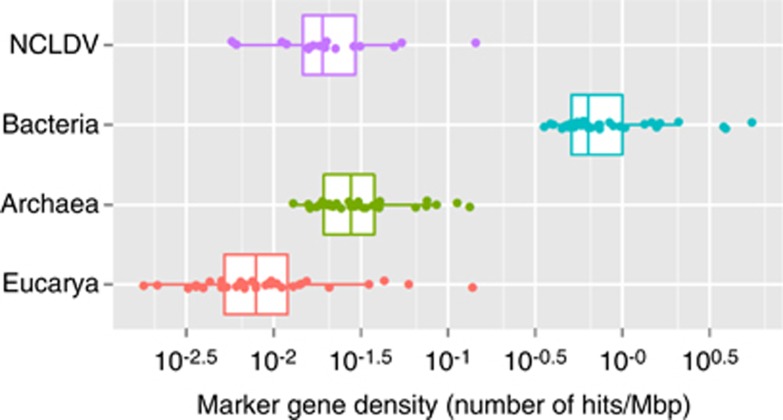
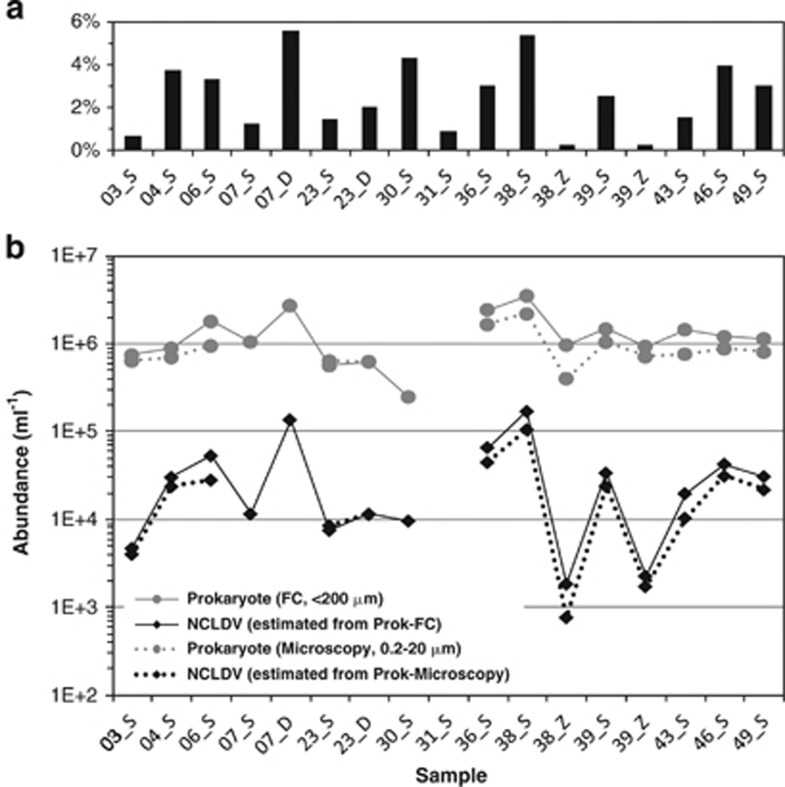
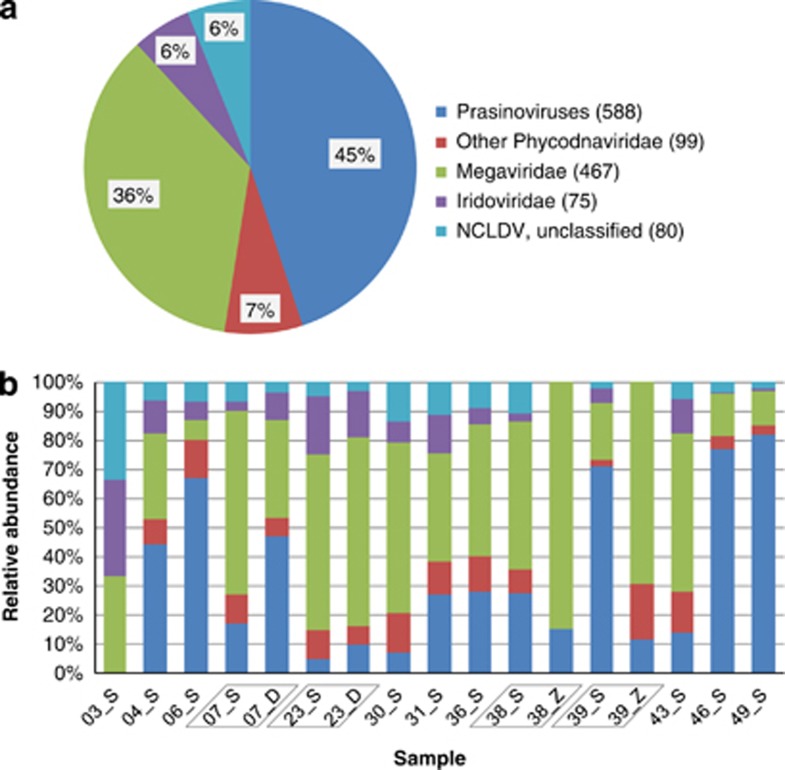
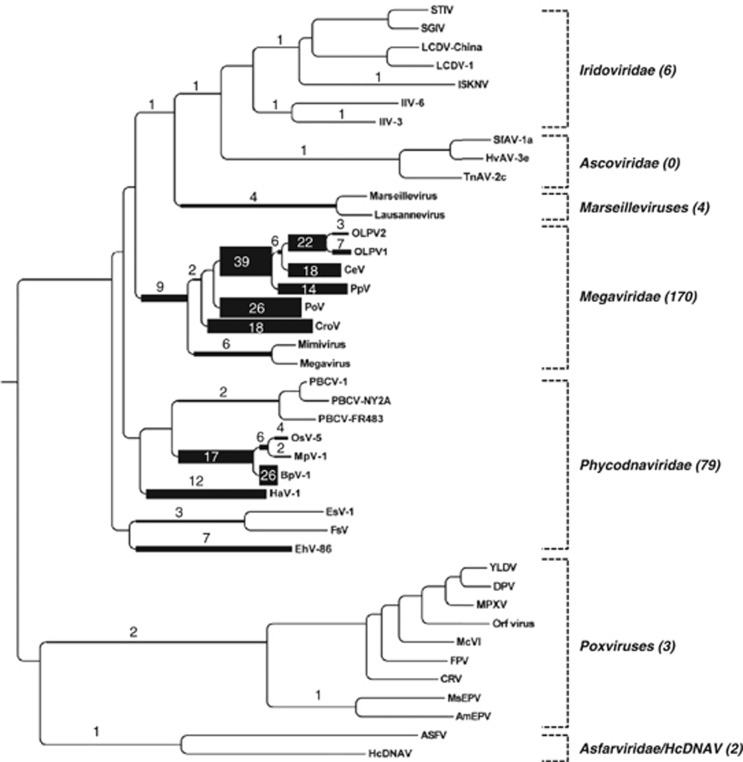
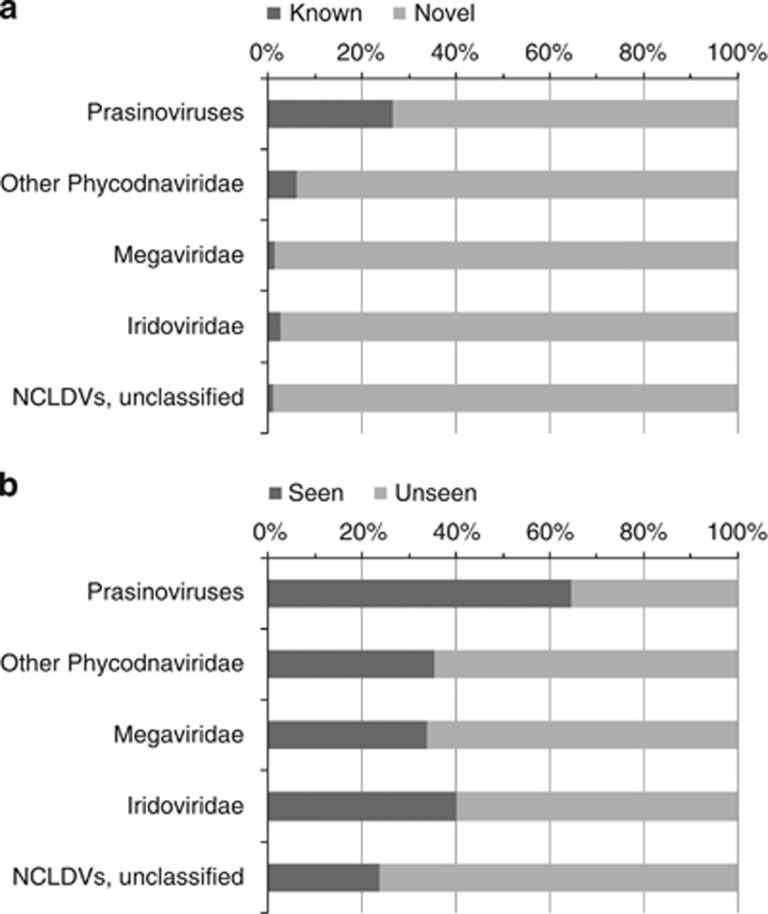
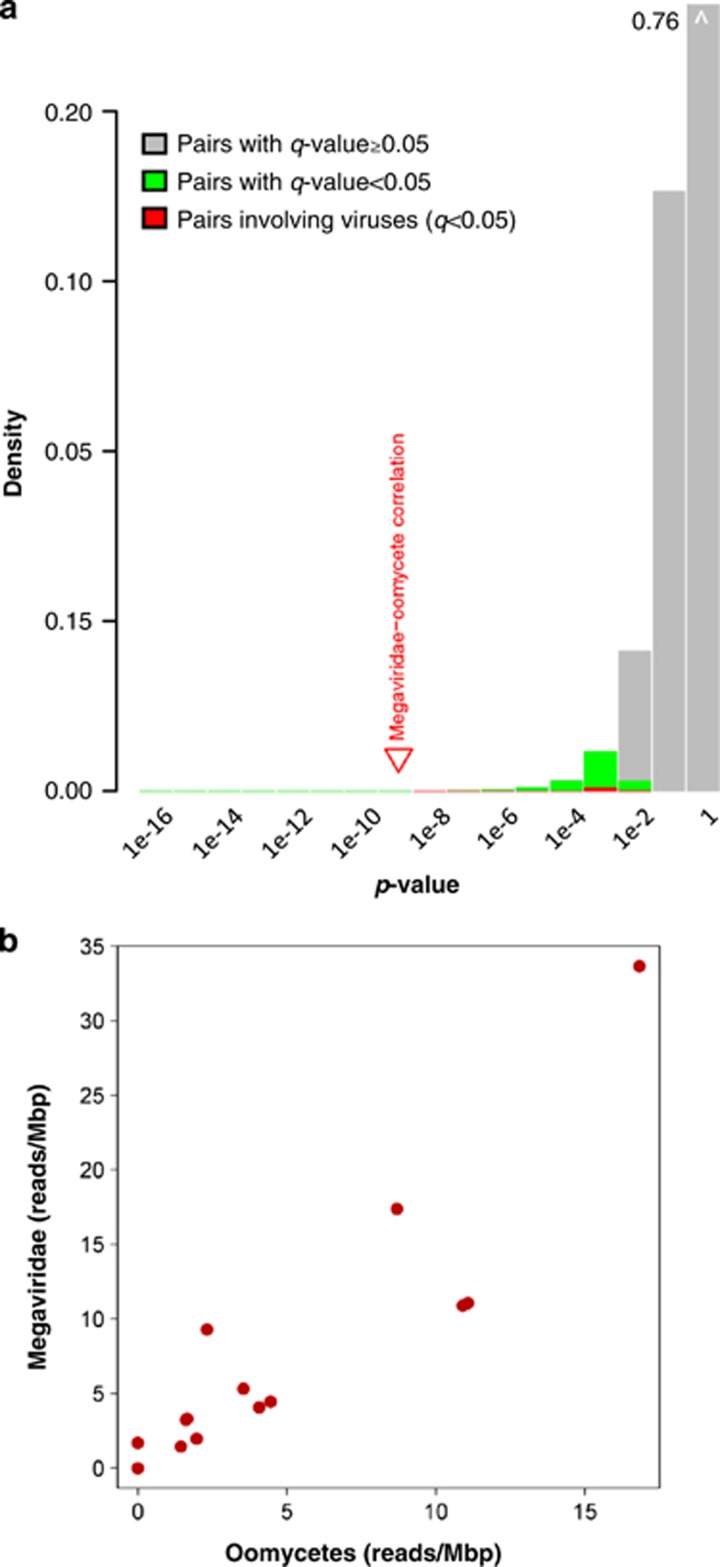
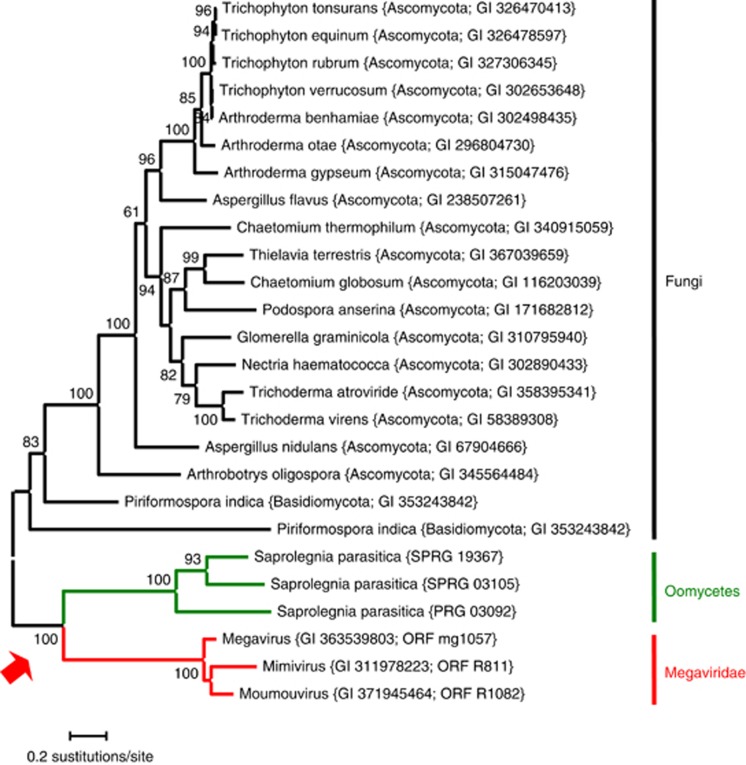
Nucleo-cytoplasmic large DNA viruses (NCLDVs) constitute a group of eukaryotic viruses that can have crucial ecological roles in the sea by accelerating the turnover of their unicellular hosts or by causing diseases in animals. To better characterize the diversity, abundance and biogeography of marine NCLDVs, we analyzed 17 metagenomes derived from microbial samples (0.2-1.6 μm size range) collected during the Tara Oceans Expedition. The sample set includes ecosystems under-represented in previous studies, such as the Arabian Sea oxygen minimum zone (OMZ) and Indian Ocean lagoons. By combining computationally derived relative abundance and direct prokaryote cell counts, the abundance of NCLDVs was found to be in the order of 10(4)-10(5) genomes ml(-1) for the samples from the photic zone and 10(2)-10(3) genomes ml(-1) for the OMZ. The Megaviridae and Phycodnaviridae dominated the NCLDV populations in the metagenomes, although most of the reads classified in these families showed large divergence from known viral genomes. Our taxon co-occurrence analysis revealed a potential association between viruses of the Megaviridae family and eukaryotes related to oomycetes. In support of this predicted association, we identified six cases of lateral gene transfer between Megaviridae and oomycetes. Our results suggest that marine NCLDVs probably outnumber eukaryotic organisms in the photic layer (per given water mass) and that metagenomic sequence analyses promise to shed new light on the biodiversity of marine viruses and their interactions with potential hosts.
Figures
References
-
- Bellec L, Grimsley N, Derelle E, Moreau H, Desdevises Y. Abundance, spatial distribution and genetic diversity of Ostreococcus tauri viruses in two different environments. Environ Microbiol Rep. 2010;2:313–321. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
