

Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue
- PMID: 23576437
- PMCID: PMC3656275
- DOI: 10.1074/jbc.M113.458414
Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue
Abstract
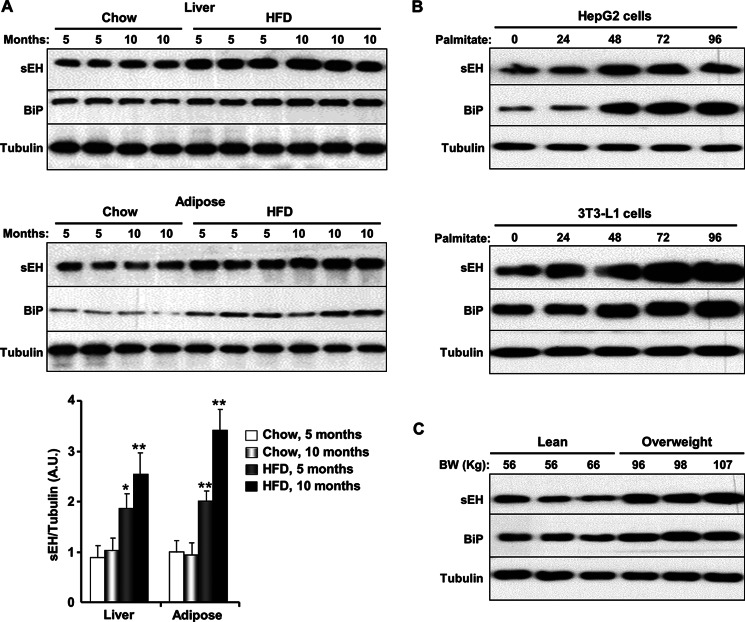
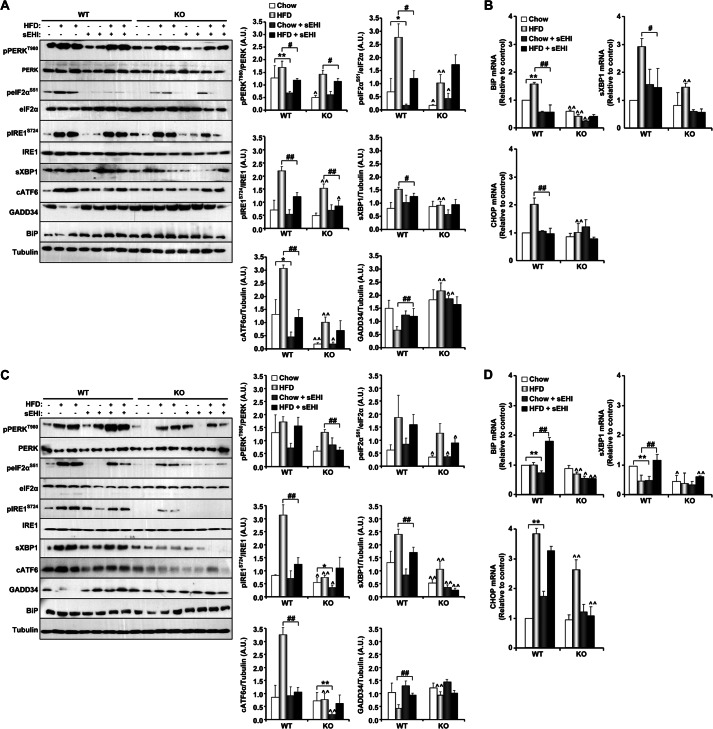
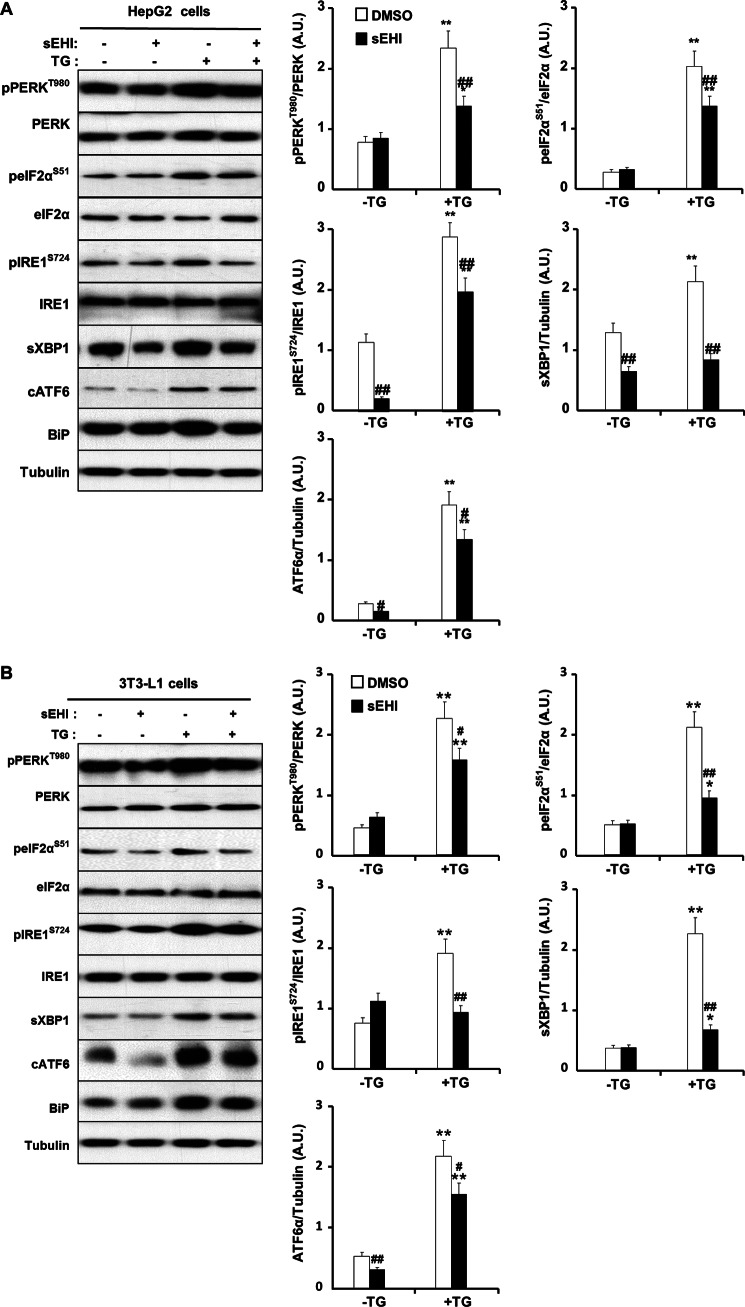
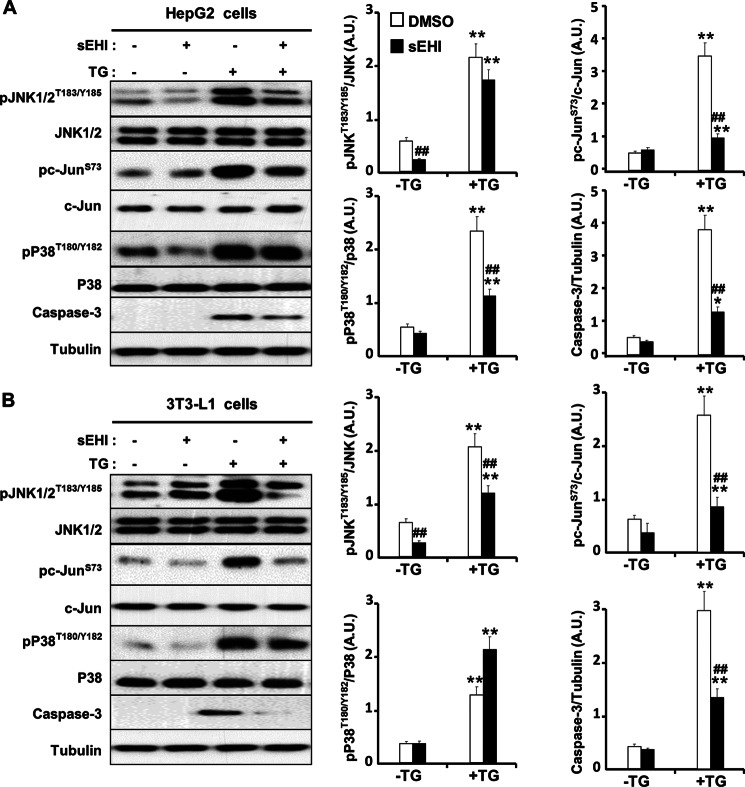
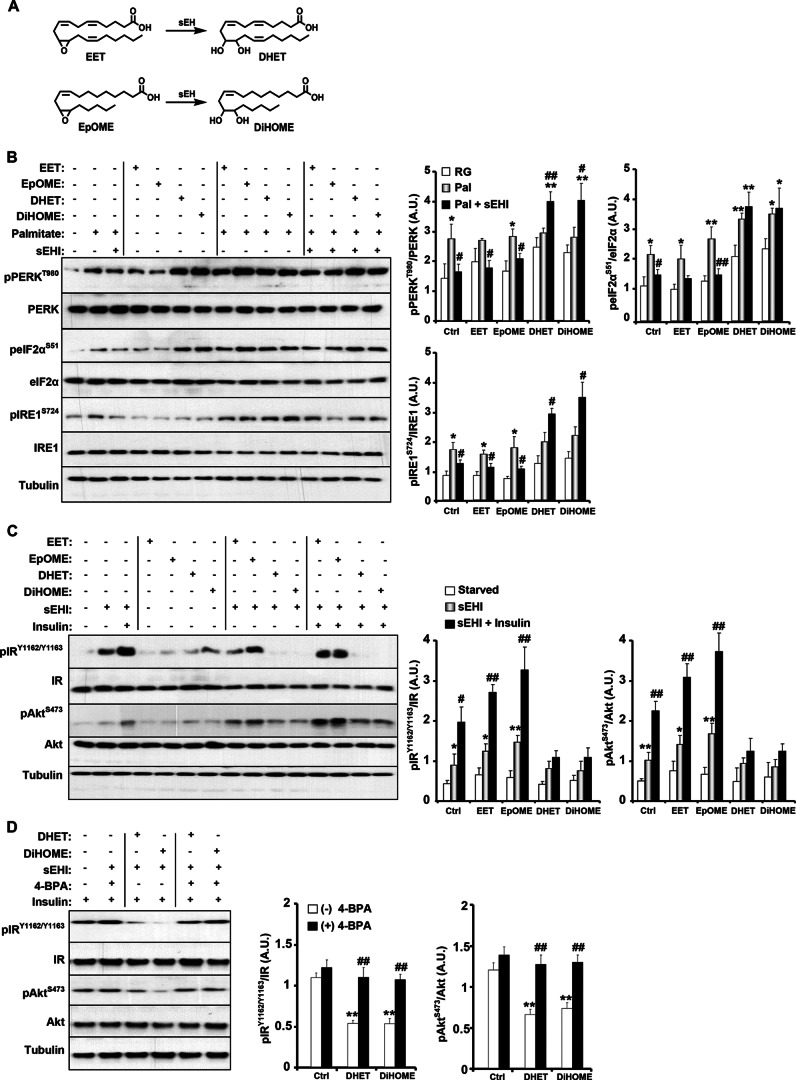
Soluble epoxide hydrolase (sEH) is a cytosolic enzyme whose inhibition has beneficial effects in cardiovascular, inflammatory, and metabolic diseases in murine models. Mice with targeted deletion or pharmacological inhibition of sEH exhibit improved insulin signaling in liver and adipose tissue. Herein, we assessed the role of sEH in regulating endoplasmic reticulum (ER) stress in liver and adipose tissue. We report that sEH expression was increased in the livers and adipose tissue of mice fed a high fat diet, the adipose tissue of overweight humans, and palmitate-treated cells. Importantly, sEH deficiency or inhibition in mice attenuated chronic high fat diet-induced ER stress in liver and adipose tissue. Similarly, pharmacological inhibition of sEH in HepG2 cells and 3T3-L1 adipocytes mitigated chemical-induced ER stress and activation of JNK, p38, and cell death. In addition, insulin signaling was enhanced in HepG2 cells treated with sEH substrates and attenuated in cells treated with sEH products. In summary, these findings demonstrate that sEH is a physiological modulator of ER stress and a potential target for mitigating complications associated with obesity.
Keywords: Endoplasmic Reticulum Stress; Epoxyeicosatrienoic Acids (EETs); Hydrolases; Inflammation; Insulin Resistance; Metabolic Regulation; Soluble Epoxide Hydrolase.
Figures
References
-
- Friedman J. M. (2000) Obesity in the new millennium. Nature 404, 632–634 - PubMed
-
- Spiegelman B. M., Flier J. S. (2001) Obesity and the regulation of energy balance. Cell 104, 531–543 - PubMed
-
- Mensah G. A., Mokdad A. H., Ford E., Narayan K. M., Giles W. H., Vinicor F., Deedwania P. C. (2004) Obesity, metabolic syndrome, and type 2 diabetes: emerging epidemics and their cardiovascular implications. Cardiol. Clin. 22, 485–504 - PubMed
-
- Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 - PubMed
-
- Hummasti S., Hotamisligil G. S. (2010) Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 107, 579–591 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
