

Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer
- PMID: 23576557
- PMCID: PMC3674118
- DOI: 10.1158/0008-5472.CAN-12-4047
Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer
Abstract
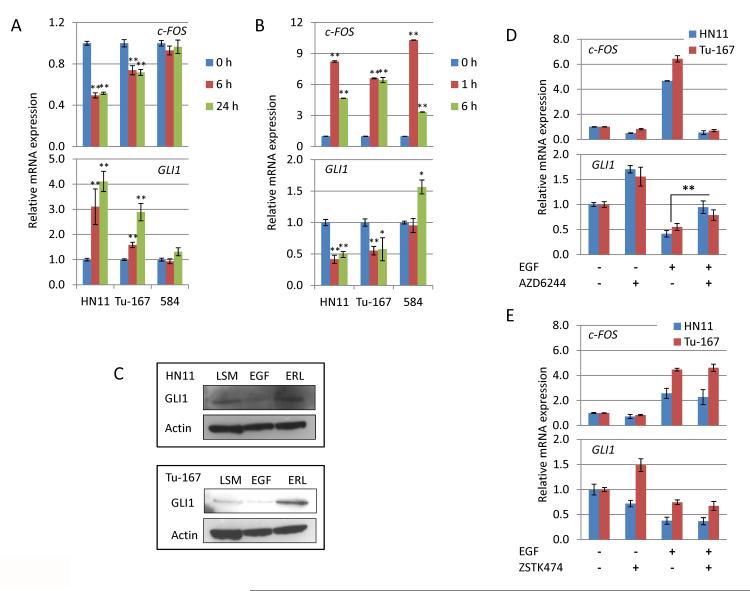
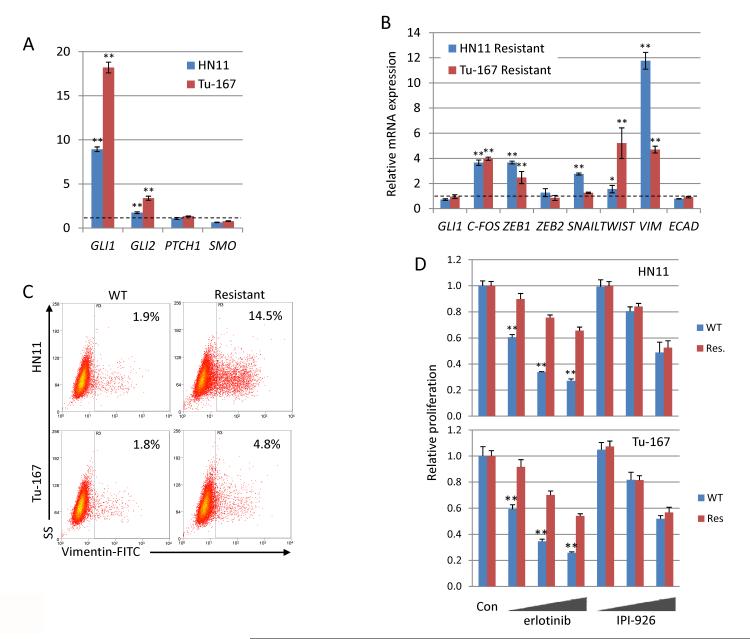
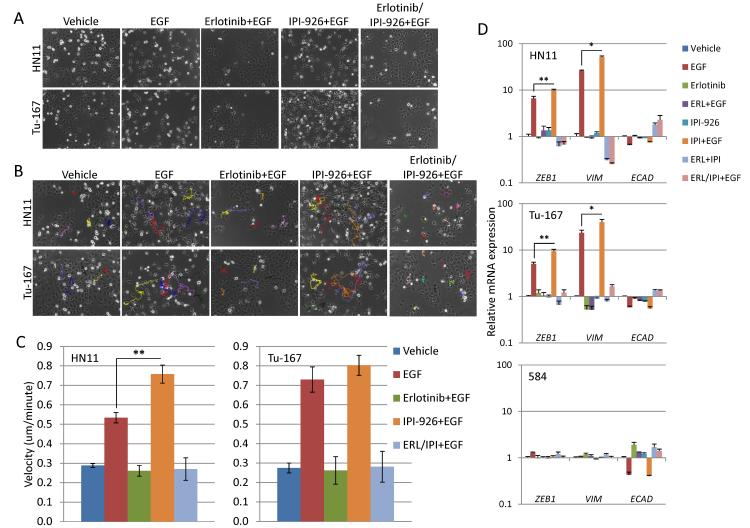
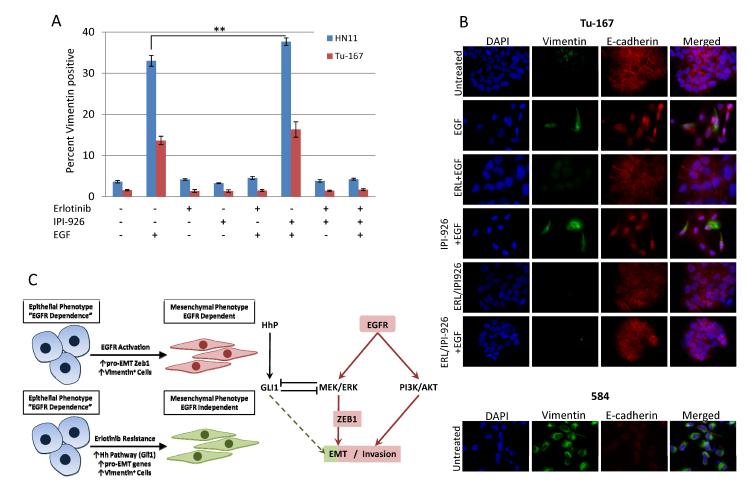
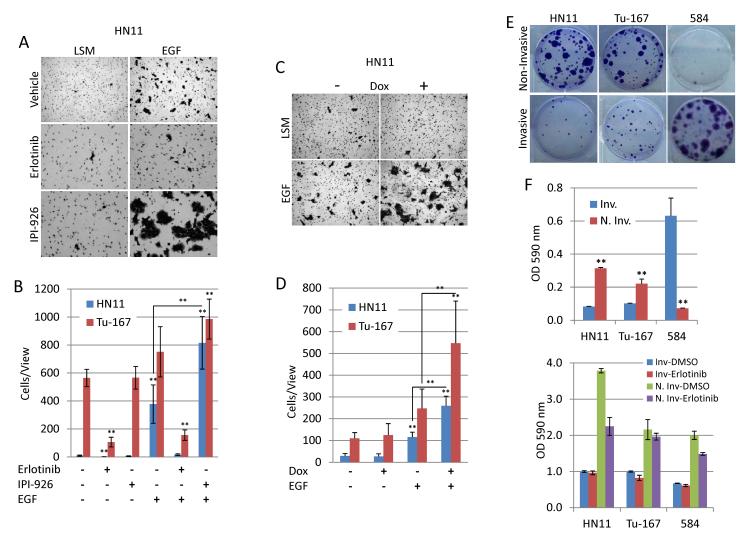
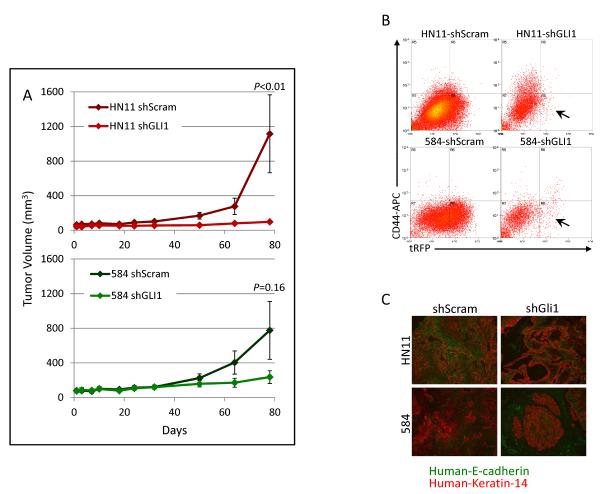
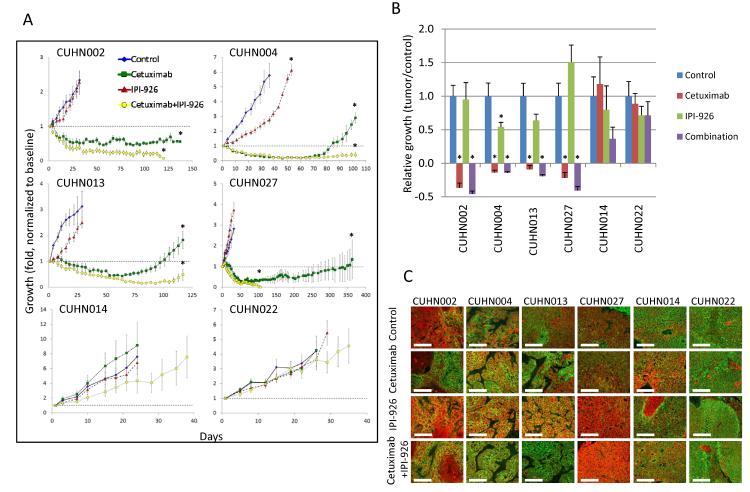
The EGF receptor (EGFR)-directed monoclonal antibody cetuximab is the only targeted therapy approved for the treatment of squamous cell carcinoma of the head and neck (HNSCC) but is only effective in a minority of patients. Epithelial-to-mesenchymal transition (EMT) has been implicated as a drug resistance mechanism in multiple cancers, and the EGFR and Hedgehog pathways (HhP) are relevant to this process, but the interplay between the two pathways has not been defined in HNSCC. Here, we show that HNSCC cells that were naturally sensitive to EGFR inhibition over time developed increased expression of the HhP transcription factor GLI1 as they became resistant after long-term EGFR inhibitor exposure. This robustly correlated with an increase in vimentin expression. Conversely, the HhP negatively regulated an EGFR-dependent, EMT-like state in HNSCC cells, and pharmacologic or genetic inhibition of HhP signaling pushed cells further into an EGFR-dependent phenotype, increasing expression of ZEB1 and VIM. In vivo treatment with cetuximab resulted in tumor shrinkage in four of six HNSCC patient-derived xenografts; however, they eventually regrew. Cetuximab in combination with the HhP inhibitor IPI-926 eliminated tumors in two cases and significantly delayed regrowth in the other two cases. Expression of EMT genes TWIST and ZEB2 was increased in sensitive xenografts, suggesting a possible resistant mesenchymal population. In summary, we report that EGFR-dependent HNSCC cells can undergo both EGFR-dependent and -independent EMT and HhP signaling is a regulator in both processes. Cetuximab plus IPI-926 forces tumor cells into an EGFR-dependent state, delaying or completely blocking tumor recurrence.
©2013 AACR.
Figures
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. - PubMed
-
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England journal of medicine. 2006;354:567–78. - PubMed
-
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. The New England journal of medicine. 2008;359:1116–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
