

Molecular events underlying Parkinson's disease - an interwoven tapestry
- PMID: 23580245
- PMCID: PMC3619247
- DOI: 10.3389/fneur.2013.00033
Molecular events underlying Parkinson's disease - an interwoven tapestry
Abstract
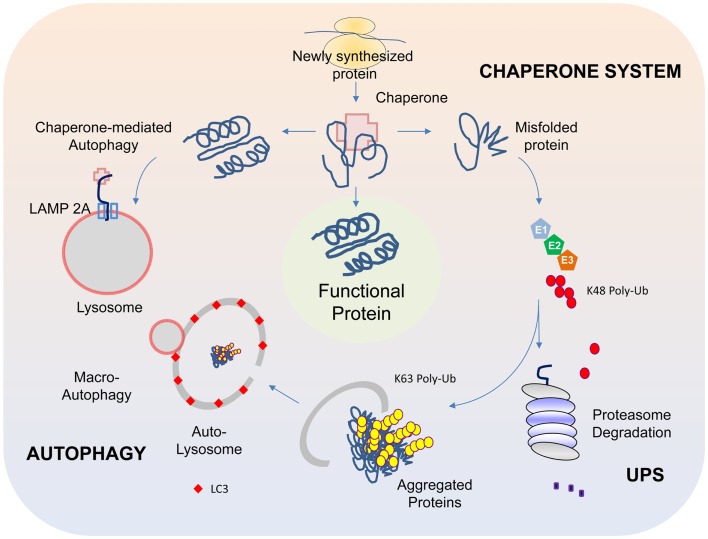
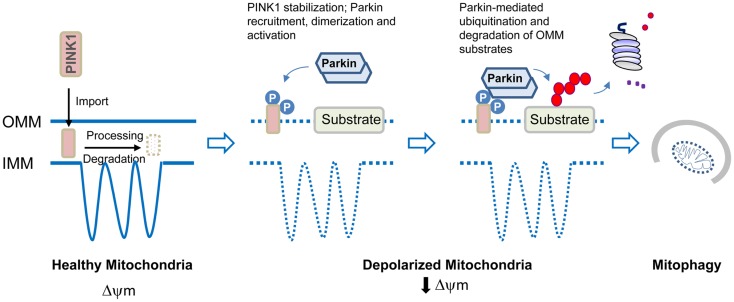
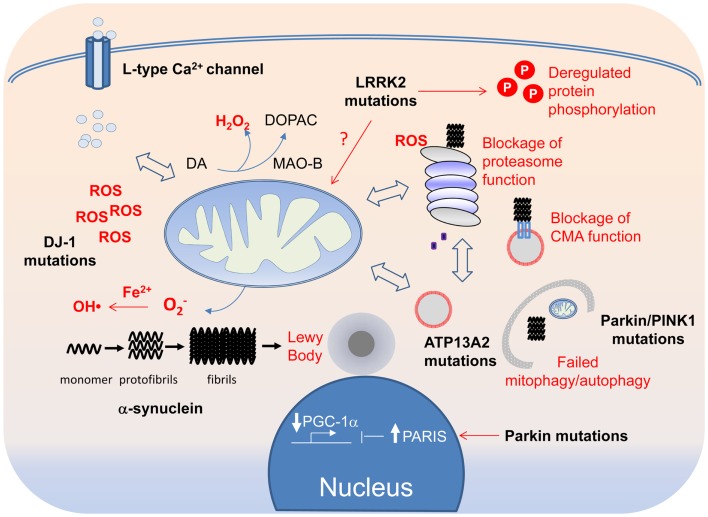
Although a subject of intense research, the mechanisms underlying dopaminergic neurodegeneration in Parkinson's disease (PD) remains poorly understood. However, a broad range of studies conducted over the past few decades, including epidemiological, genetic, and post-mortem analysis, as well as in vitro and in vivo modeling, have contributed significantly to our understanding of the pathogenesis of the disease. In particular, the recent identification and functional characterization of several genes, including α-synuclein, parkin, DJ-1, PINK1, and LRRK2, whose mutations are causative of rare familial forms of PD have provided tremendous insights into the molecular pathways underlying dopaminergic neurodegeneration. Collectively, these studies implicate aberrant mitochondrial and protein homeostasis as key contributors to the development of PD, with oxidative stress likely acting as an important nexus between the two pathogenic events. Aberrations in homeostatic processes leading to protein aggregation and mitochondrial dysfunction may arise intrinsically in substantia nigra pars compacta dopaminergic neurons as a result of impairments in the ubiquitin-proteasome system, failure in autophagy-mediated clearance, alterations of mitochondrial dynamics, redox imbalance, iron mishandling, dopamine dysregulation, or simply from the chronic pace-making activity of nigra-localized L-type calcium channels, or extrinsically from non-autonomous sources of stress. Given the myriad of culprits implicated, the pathogenesis of PD necessarily involves an intricate network of interwoven pathways rather than a linear sequence of events. Obviously, understanding how the various disease-associated pathways interact with and influence each other is of mechanistic and therapeutic importance. Here, we shall discuss some key PD-related pathways and how they are interwoven together into a tapestry of events.
Keywords: Parkinson disease; autophagy; mitophagy; oxidative stress; proteasome; protein aggregation.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous