

Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing
- PMID: 23580730
- PMCID: PMC3668365
- DOI: 10.1101/gr.150342.112
Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing
Abstract
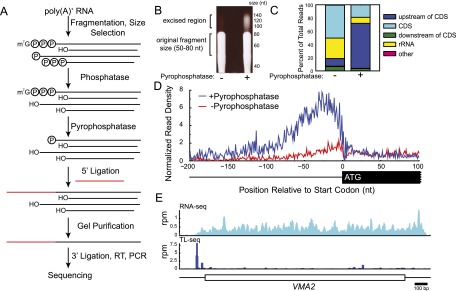
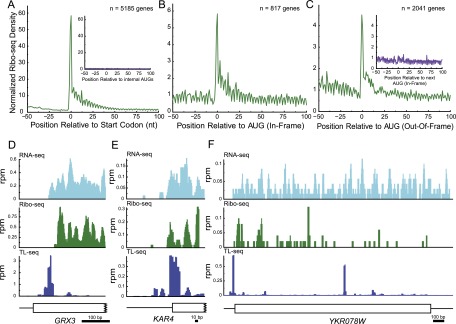
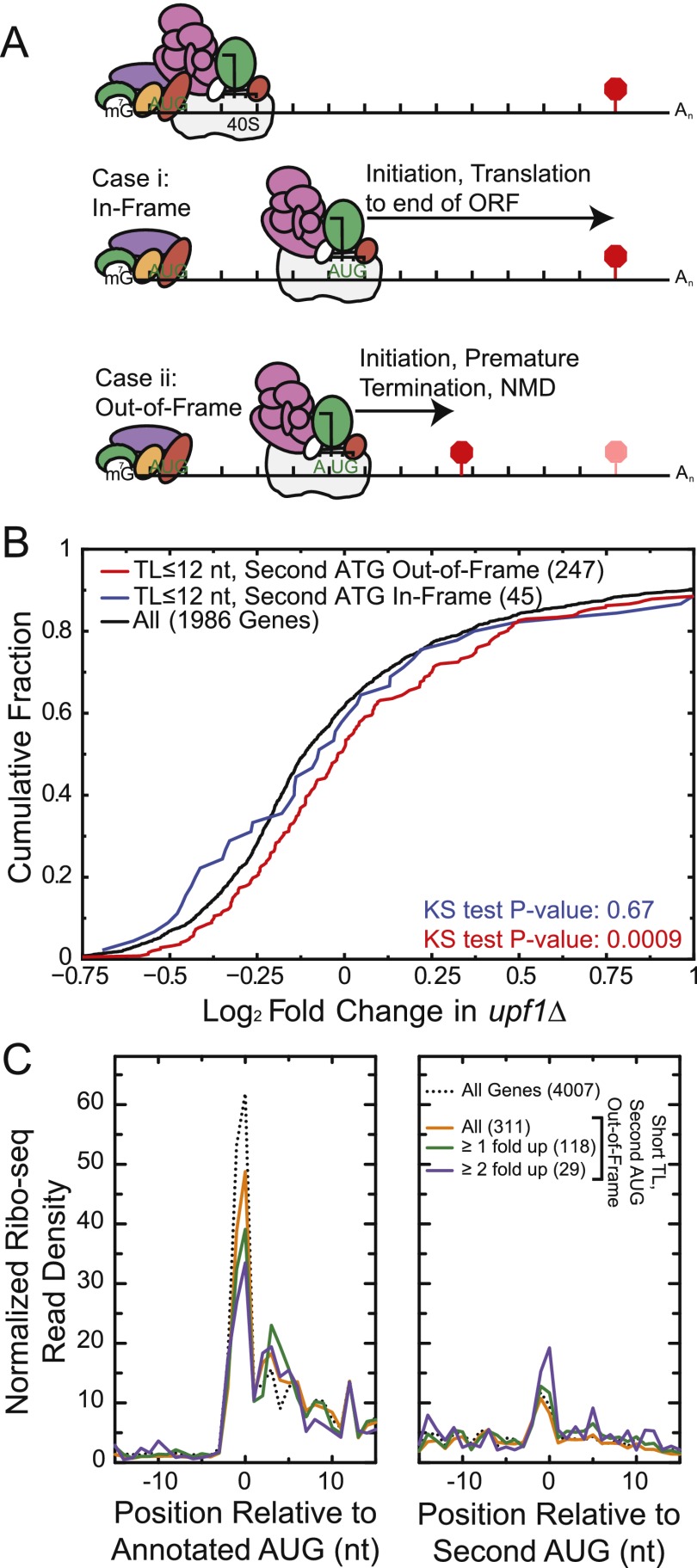
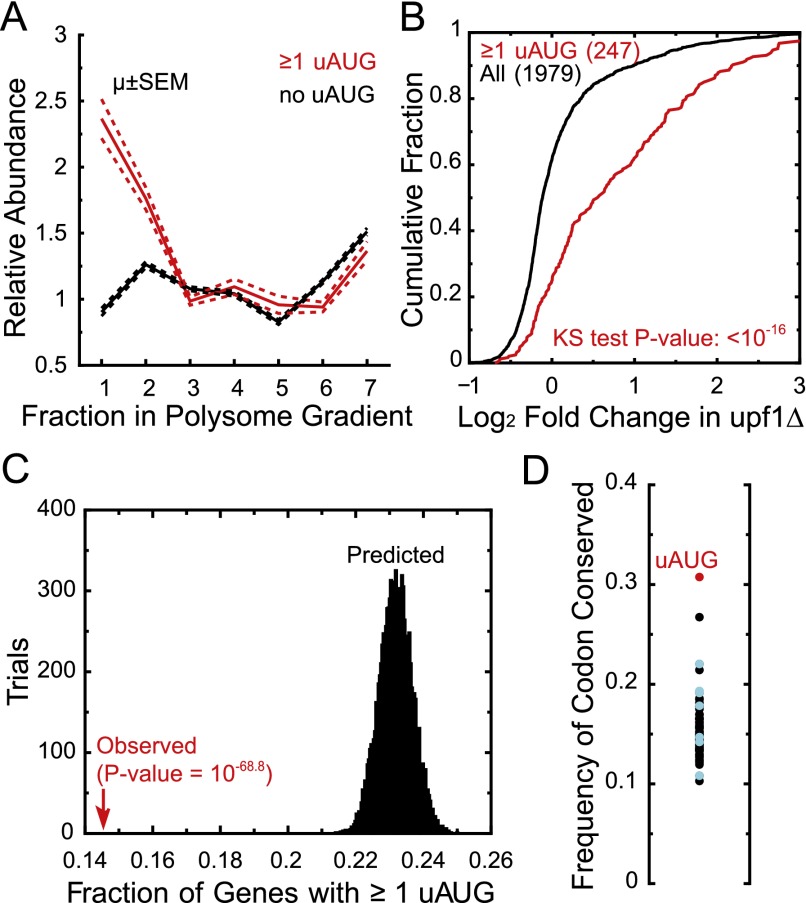
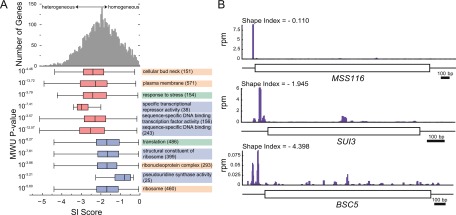
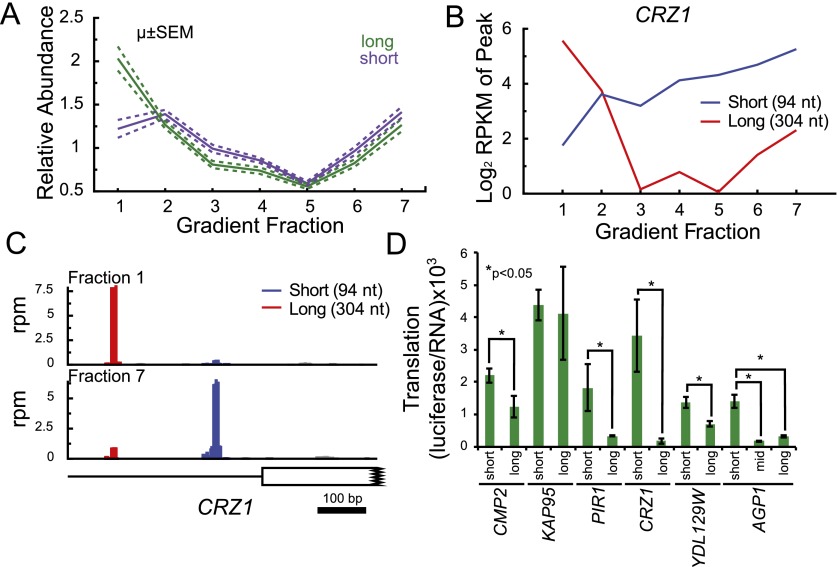
Transcript leaders (TLs) can have profound effects on mRNA translation and stability. To map TL boundaries genome-wide, we developed TL-sequencing (TL-seq), a technique combining enzymatic capture of m(7)G-capped mRNA 5' ends with high-throughput sequencing. TL-seq identified mRNA start sites for the majority of yeast genes and revealed many examples of intragenic TL heterogeneity. Surprisingly, TL-seq identified transcription initiation sites within 6% of protein-coding regions, and these sites were concentrated near the 5' ends of ORFs. Furthermore, ribosome density analysis showed these truncated mRNAs are translated. Translation-associated TL-seq (TATL-seq), which combines TL-seq with polysome fractionation, enabled annotation of TLs, and simultaneously assayed their function in translation. Using TATL-seq to address relationships between TL features and translation of the downstream ORF, we observed that upstream AUGs (uAUGs), and no other upstream codons, were associated with poor translation and nonsense-mediated mRNA decay (NMD). We also identified hundreds of genes with very short TLs, and demonstrated that short TLs were associated with poor translation initiation at the annotated start codon and increased initiation at downstream AUGs. This frequently resulted in out-of-frame translation and subsequent termination at premature termination codons, culminating in NMD of the transcript. Unlike previous approaches, our technique enabled observation of alternative TL variants for hundreds of genes and revealed significant differences in translation in genes with distinct TL isoforms. TL-seq and TATL-seq are useful tools for annotation and functional characterization of TLs, and can be applied to any eukaryotic system to investigate TL-mediated regulation of gene expression.
Figures
References
-
- Aitken CE, Lorsch JR 2012. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19: 568–576 - PubMed
-
- Altschul SF, Erickson BW 1985. Significance of nucleotide sequence alignments: A method for random sequence permutation that preserves dinucleotide and codon usage. Mol Biol Evol 2: 526–538 - PubMed
-
- Beltzer JP, Chang LF, Hinkkanen AE, Kohlhaw GB 1986. Structure of yeast LEU4. The 5′ flanking region contains features that predict two modes of control and two productive translation starts. J Biol Chem 261: 5160–5167 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases