

Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver
- PMID: 23583108
- PMCID: PMC3966570
- DOI: 10.1016/j.neuron.2013.02.011
Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver
Abstract
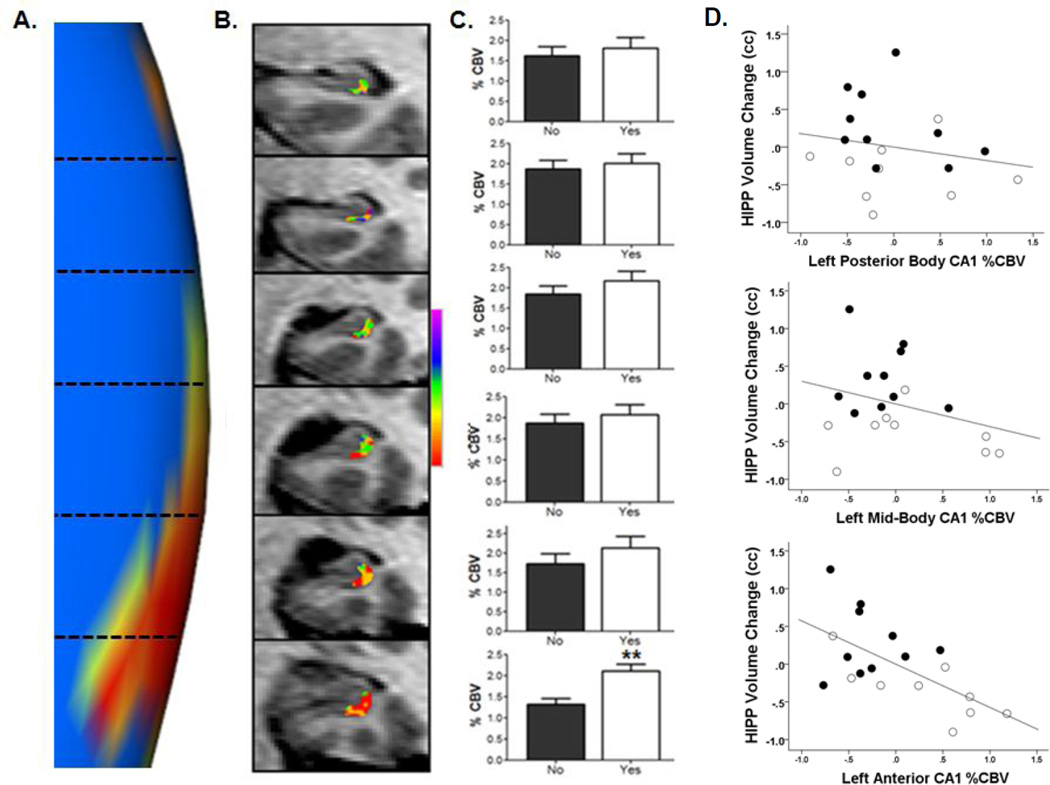
The hippocampus in schizophrenia is characterized by both hypermetabolism and reduced size. It remains unknown whether these abnormalities are mechanistically linked. Here we addressed this question by using MRI tools that can map hippocampal metabolism and structure in patients and mouse models. In at-risk patients, hypermetabolism was found to begin in CA1 and spread to the subiculum after psychosis onset. CA1 hypermetabolism at baseline predicted hippocampal atrophy, which occurred during progression to psychosis, most prominently in similar regions. Next, we used ketamine to model conditions of acute psychosis in mice. Acute ketamine reproduced a similar regional pattern of hypermetabolism, while repeated exposure shifted the hippocampus to a hypermetabolic basal state with concurrent atrophy and pathology in parvalbumin-expressing interneurons. Parallel in vivo experiments using the glutamate-reducing drug LY379268 and direct measurements of extracellular glutamate showed that glutamate drives both neuroimaging abnormalities. These findings show that hippocampal hypermetabolism leads to atrophy in psychotic disorder and suggest glutamate as a pathogenic driver.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures






Comment in
-
A mechanistic approach to preventing schizophrenia in at-risk individuals.Neuron. 2013 Apr 10;78(1):1-3. doi: 10.1016/j.neuron.2013.03.024. Neuron. 2013. PMID: 23583102
References
-
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. - PubMed
-
- Benes FM. Evidence for altered trisynaptic circuitry in schizophrenic hippocampus. Biological psychiatry. 1999;46:589–599. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P40HD03110/HD/NICHD NIH HHS/United States
- R21MH086125/MH/NIMH NIH HHS/United States
- U54EB005149/EB/NIBIB NIH HHS/United States
- 1R01MH093398-01/MH/NIMH NIH HHS/United States
- K23MH066279/MH/NIMH NIH HHS/United States
- P50 AG008702/AG/NIA NIH HHS/United States
- K23MH090563/MH/NIMH NIH HHS/United States
- UL1 RR024156/RR/NCRR NIH HHS/United States
- K23 MH066279/MH/NIMH NIH HHS/United States
- K23 MH090563/MH/NIMH NIH HHS/United States
- K23 AG000946/AG/NIA NIH HHS/United States
- UL1TR000040/TR/NCATS NIH HHS/United States
- P50MH086385/MH/NIMH NIH HHS/United States
- UL1 TR000040/TR/NCATS NIH HHS/United States
- UL1RR024156/RR/NCRR NIH HHS/United States
- P50 MH086385/MH/NIMH NIH HHS/United States
- R01 MH093398/MH/NIMH NIH HHS/United States
- R01 AG025161/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
