

Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas
- PMID: 23583981
- PMCID: PMC3727232
- DOI: 10.1038/ng.2611
Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas
Abstract
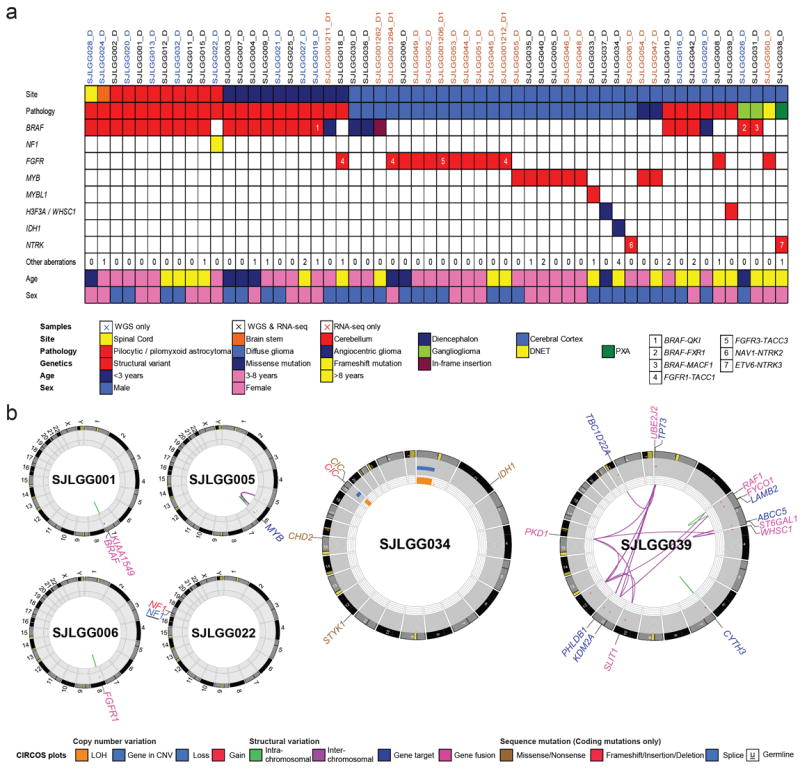
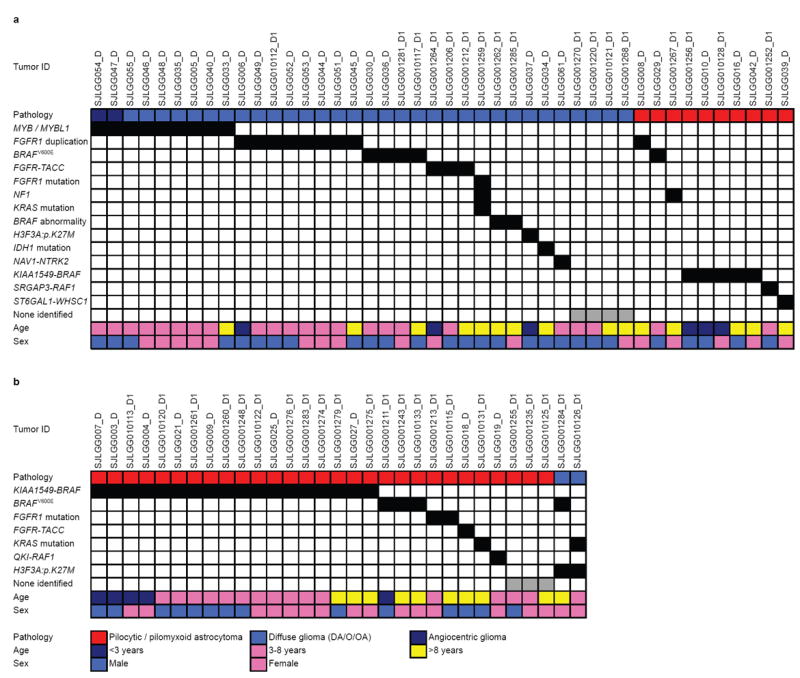
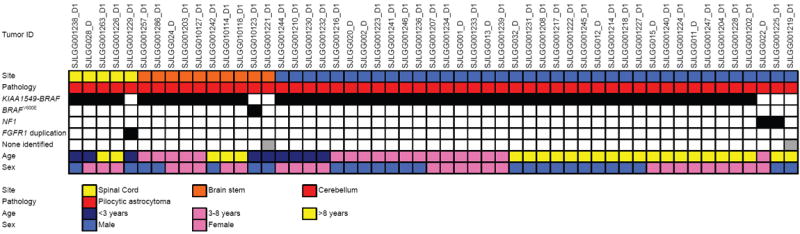
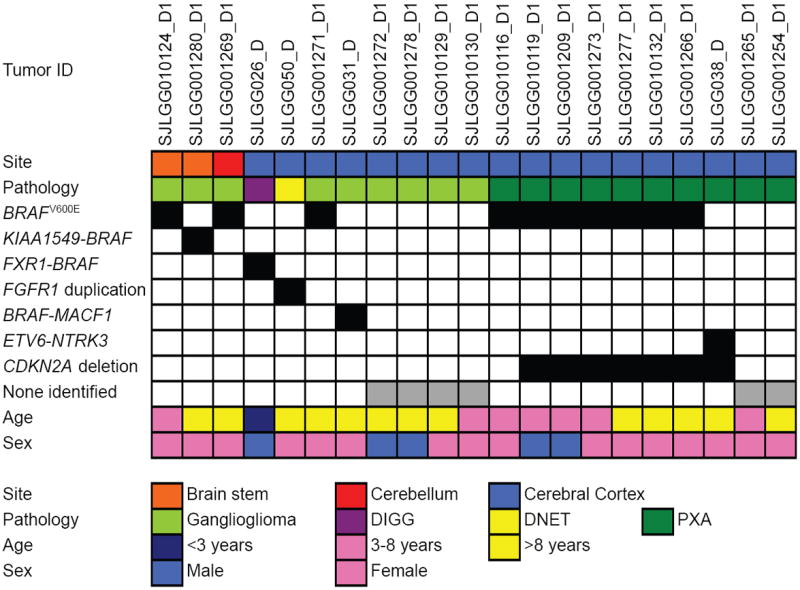
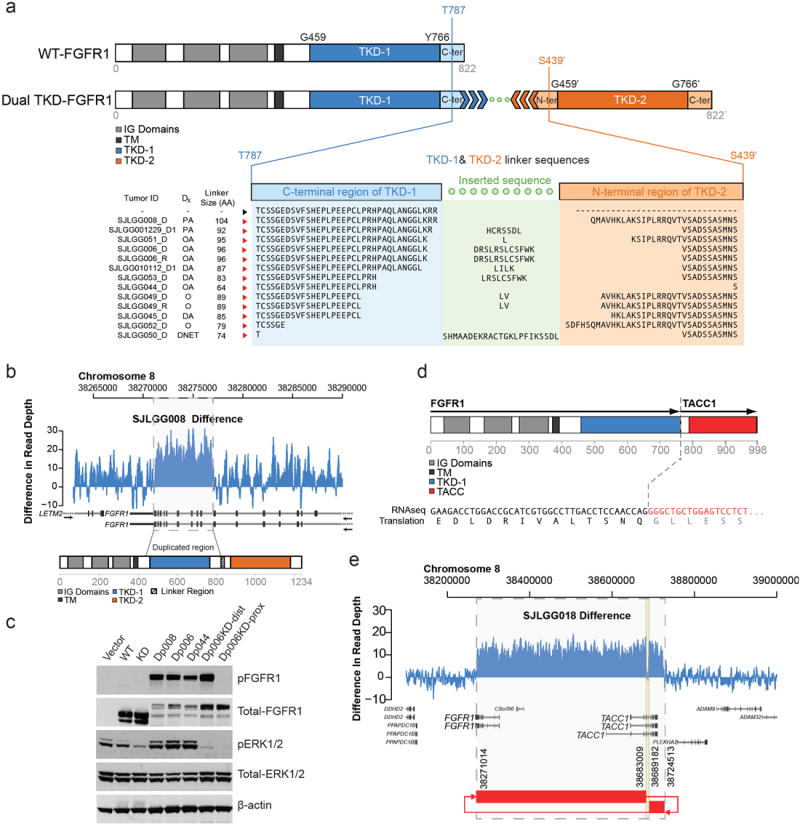
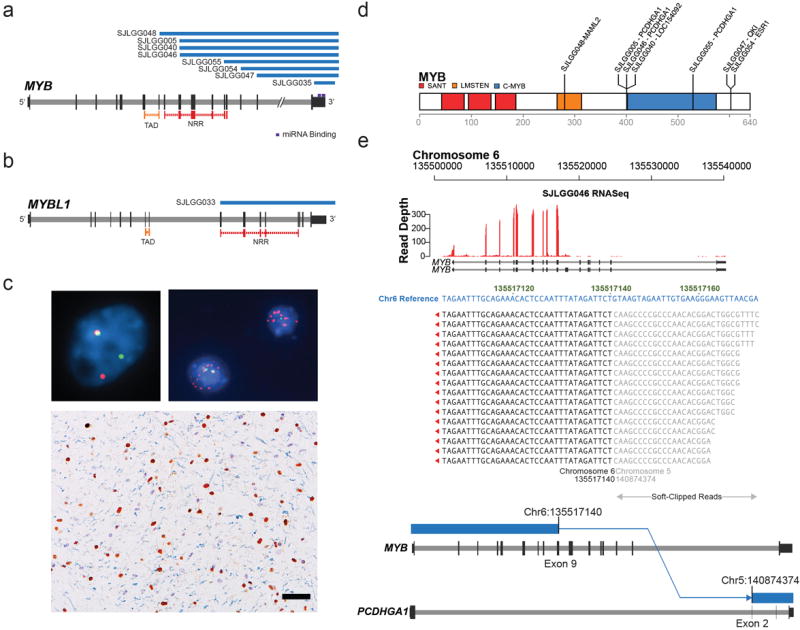
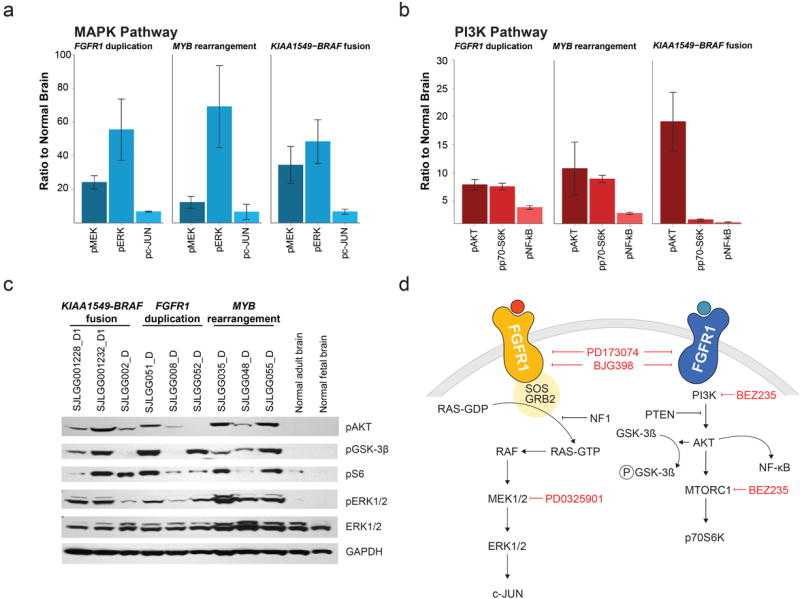
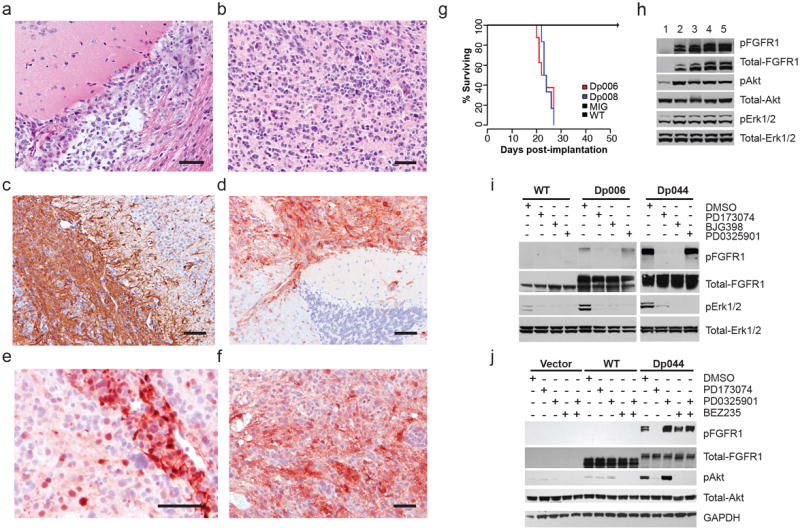
The most common pediatric brain tumors are low-grade gliomas (LGGs). We used whole-genome sequencing to identify multiple new genetic alterations involving BRAF, RAF1, FGFR1, MYB, MYBL1 and genes with histone-related functions, including H3F3A and ATRX, in 39 LGGs and low-grade glioneuronal tumors (LGGNTs). Only a single non-silent somatic alteration was detected in 24 of 39 (62%) tumors. Intragenic duplications of the portion of FGFR1 encoding the tyrosine kinase domain (TKD) and rearrangements of MYB were recurrent and mutually exclusive in 53% of grade II diffuse LGGs. Transplantation of Trp53-null neonatal astrocytes expressing FGFR1 with the duplication involving the TKD into the brains of nude mice generated high-grade astrocytomas with short latency and 100% penetrance. FGFR1 with the duplication induced FGFR1 autophosphorylation and upregulation of the MAPK/ERK and PI3K pathways, which could be blocked by specific inhibitors. Focusing on the therapeutically challenging diffuse LGGs, our study of 151 tumors has discovered genetic alterations and potential therapeutic targets across the entire range of pediatric LGGs and LGGNTs.
Figures
References
-
- Bouffet E, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:1358–63. - PubMed
-
- Duffner PK, Cohen ME, Myers MH, Heise HW. Survival of children with brain tumors: SEER Program, 1973-1980. Neurology. 1986;36:597–601. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
