

Accurate detection of differential RNA processing
- PMID: 23585274
- PMCID: PMC3664801
- DOI: 10.1093/nar/gkt211
Accurate detection of differential RNA processing
Abstract
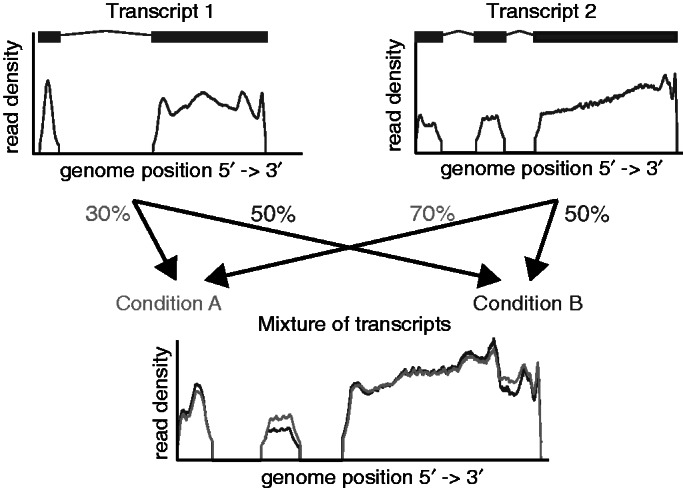
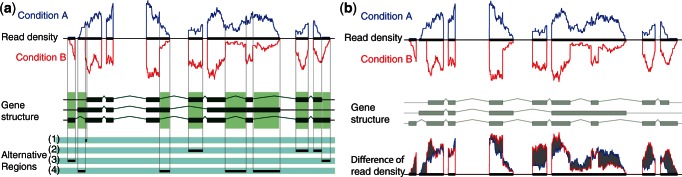
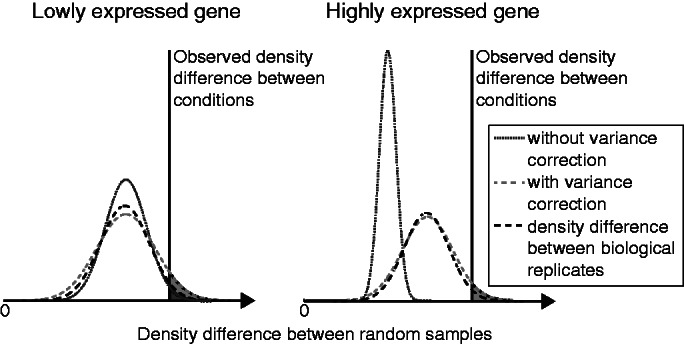
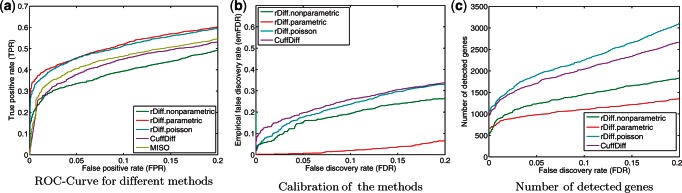
Deep transcriptome sequencing (RNA-Seq) has become a vital tool for studying the state of cells in the context of varying environments, genotypes and other factors. RNA-Seq profiling data enable identification of novel isoforms, quantification of known isoforms and detection of changes in transcriptional or RNA-processing activity. Existing approaches to detect differential isoform abundance between samples either require a complete isoform annotation or fall short in providing statistically robust and calibrated significance estimates. Here, we propose a suite of statistical tests to address these open needs: a parametric test that uses known isoform annotations to detect changes in relative isoform abundance and a non-parametric test that detects differential read coverages and can be applied when isoform annotations are not available. Both methods account for the discrete nature of read counts and the inherent biological variability. We demonstrate that these tests compare favorably to previous methods, both in terms of accuracy and statistical calibrations. We use these techniques to analyze RNA-Seq libraries from Arabidopsis thaliana and Drosophila melanogaster. The identified differential RNA processing events were consistent with RT-qPCR measurements and previous studies. The proposed toolkit is available from http://bioweb.me/rdiff and enables in-depth analyses of transcriptomes, with or without available isoform annotation.
Figures
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
-
- Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. - PubMed
-
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
