

Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1
- PMID: 23585456
- PMCID: PMC3680595
- DOI: 10.1158/0008-5472.CAN-12-4619
Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1
Abstract
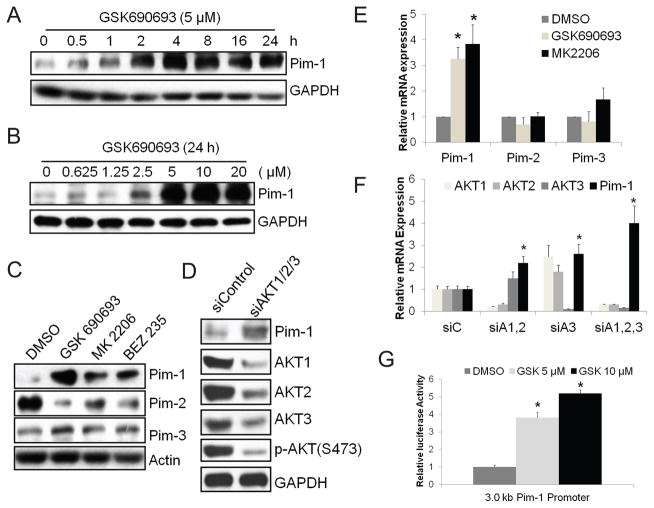
The PI3K/AKT pathway is hyperactivated in prostate cancer but its effective therapeutic targeting has proven difficult. In particular, the antitumor activity of AKT inhibitors is attenuated by upregulation of receptor tyrosine kinases (RTK) through an uncharacterized feedback mechanism. In this report, we show that RNA interference-mediated silencing or pharmacologic inhibition of Pim-1 activity curtails AKT inhibitor-induced upregulation of RTKs in prostate cancer cells. Although Pim kinases have been implicated in cap-dependent translational control, we find that in the context of AKT inhibition, the expression of RTKs is controlled by Pim-1 in a cap-independent manner by controlling internal ribosome entry. Combination of Pim and AKT inhibitors resulted in synergistic inhibition of prostate tumor growth in vitro and in vivo. Together, our results show that Pim-1 mediates resistance to AKT inhibition and suggest its targeting to improve the efficacy of AKT inhibitors in anticancer therapy.
©2013 AACR.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures





Comment in
-
Rational cotargeting of Pim-1 and Akt in prostate cancer.Expert Rev Anticancer Ther. 2013 Aug;13(8):937-9. doi: 10.1586/14737140.2013.816461. Expert Rev Anticancer Ther. 2013. PMID: 23984895
References
-
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. - PubMed
-
- Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–9. - PubMed
-
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. - PubMed
-
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
