

The "fossilized" mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate
- PMID: 23587068
- PMCID: PMC3646698
- DOI: 10.1186/1741-7007-11-29
The "fossilized" mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate
Abstract
Background: The mitochondrial genomes of flowering plants vary greatly in size, gene content, gene order, mutation rate and level of RNA editing. However, the narrow phylogenetic breadth of available genomic data has limited our ability to reconstruct these traits in the ancestral flowering plant and, therefore, to infer subsequent patterns of evolution across angiosperms.
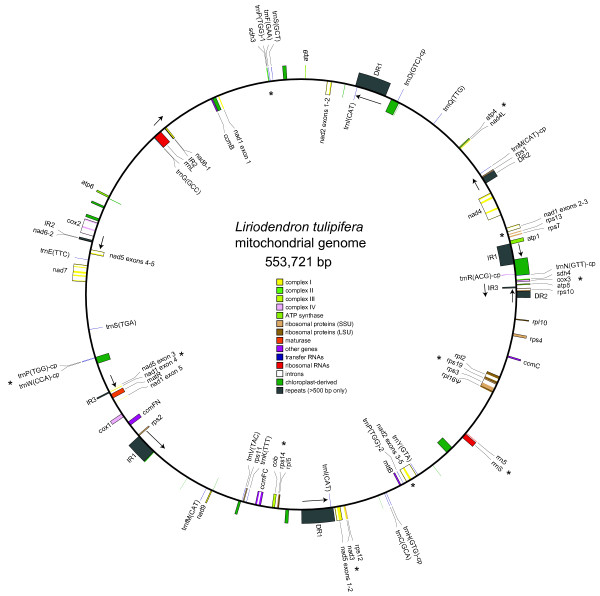
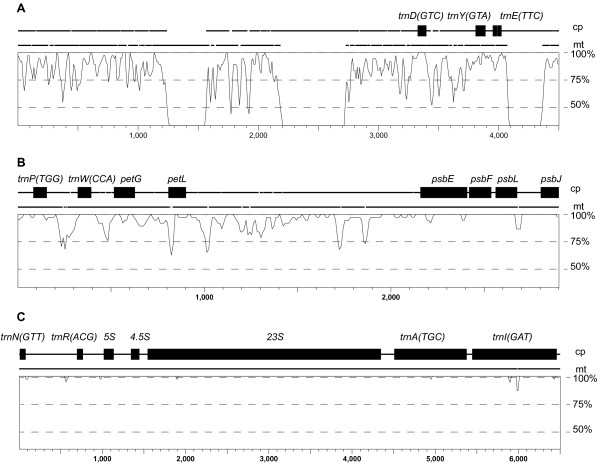
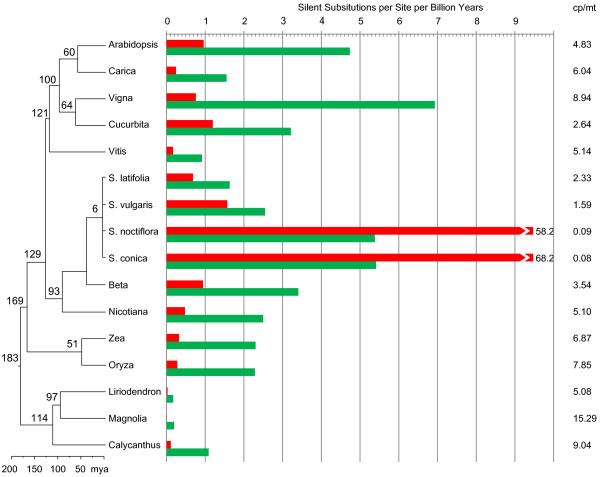
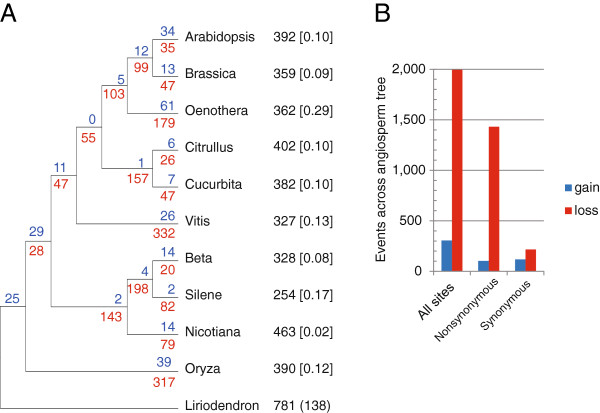
Results: We sequenced the mitochondrial genome of Liriodendron tulipifera, the first from outside the monocots or eudicots. This 553,721 bp mitochondrial genome has evolved remarkably slowly in virtually all respects, with an extraordinarily low genome-wide silent substitution rate, retention of genes frequently lost in other angiosperm lineages, and conservation of ancestral gene clusters. The mitochondrial protein genes in Liriodendron are the most heavily edited of any angiosperm characterized to date. Most of these sites are also edited in various other lineages, which allowed us to polarize losses of editing sites in other parts of the angiosperm phylogeny. Finally, we added comprehensive gene sequence data for two other magnoliids, Magnolia stellata and the more distantly related Calycanthus floridus, to measure rates of sequence evolution in Liriodendron with greater accuracy. The Magnolia genome has evolved at an even lower rate, revealing a roughly 5,000-fold range of synonymous-site divergence among angiosperms whose mitochondrial gene space has been comprehensively sequenced.
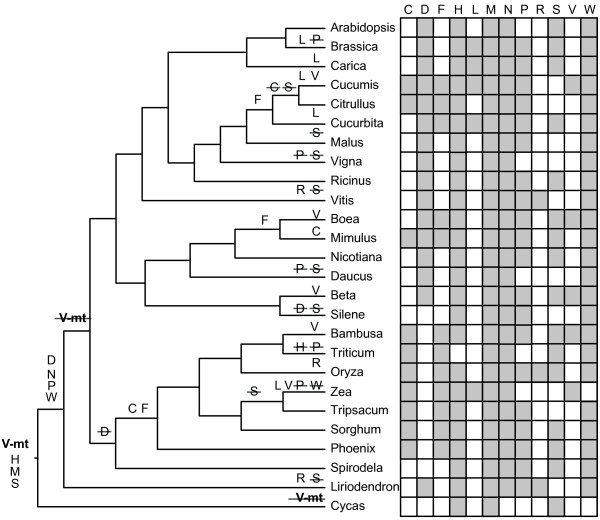
Conclusions: Using Liriodendron as a guide, we estimate that the ancestral flowering plant mitochondrial genome contained 41 protein genes, 14 tRNA genes of mitochondrial origin, as many as 7 tRNA genes of chloroplast origin, >700 sites of RNA editing, and some 14 colinear gene clusters. Many of these gene clusters, genes and RNA editing sites have been variously lost in different lineages over the course of the ensuing ∽200 million years of angiosperm evolution.
Figures

Comment in
-
Mitochondrial genomes as living 'fossils'.BMC Biol. 2013 Apr 15;11:30. doi: 10.1186/1741-7007-11-30. BMC Biol. 2013. PMID: 23587103 Free PMC article.
References
-
- Mower JP, Sloan DB, Alverson AJ. In: Plant Genome Diversity. Wendel JF, Greilhuber J, Dolezel J, Leitch IJ, editor. Vol. 1. Vienna: Springer Vienna; 2012. Plant mitochondrial genome diversity: the genomics revolution; pp. 123–144.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
