

Fool's gold, lost treasures, and the randomized clinical trial
- PMID: 23587187
- PMCID: PMC3639810
- DOI: 10.1186/1471-2407-13-193
Fool's gold, lost treasures, and the randomized clinical trial
Abstract
Background: Randomized controlled trials with a survival endpoint are the gold standard for clinical research, but have failed to achieve cures for most advanced malignancies. The high costs of randomized clinical trials slow progress (thereby causing avoidable loss of life) and increase health care costs.
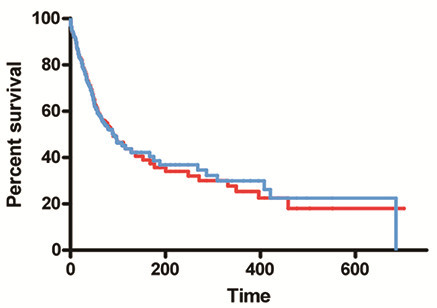
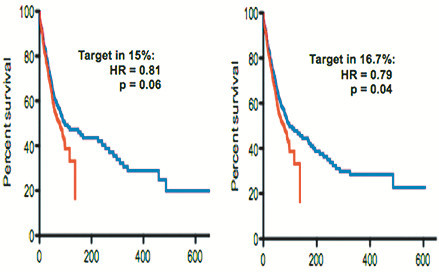
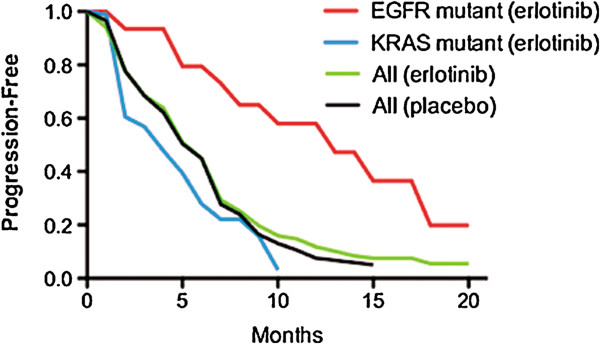
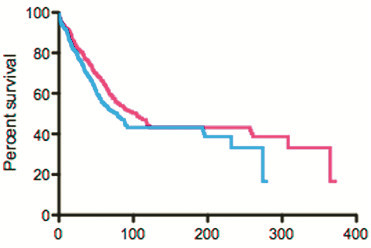
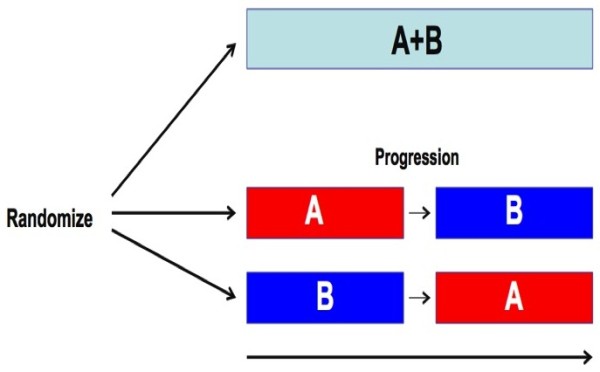
Discussion: A malignancy may be caused by several different mutations. Therapies effective vs one mutation may be discarded due to lack of statistical significance across the entire population. Conversely, expensive large randomized trials may have sufficient statistical power to demonstrate benefit despite the therapy only working in subgroups. Non-cost-effective therapy is then applied to all patients (including subgroups it cannot help). Randomized trials comparing therapies with different mechanisms of action are misleading since they may conclude the therapies are "equivalent" despite benefitting different subpopulations, or may erroneously conclude that one therapy is superior simply because it targets a larger subpopulation. Furthermore, minor variances in patient selection may determine study outcome, a therapy may be discarded as ineffective despite substantial benefit in one subpopulation if harmful in another, randomized trials may more effectively detect therapies with minor benefit in most patients vs marked benefit in subpopulations, and randomized trials in unselected patients may erroneously conclude that "shot-gun" combinations are superior to single agents when sequential administration of personalized single agents might work better and spare patients treatment with drugs that cannot help them. We must identify predictive biomarkers early by comparing responding to progressing patients in phase I-II trials. Enriching randomized trials for biomarker-positive patients can markedly reduce required patient numbers and costs despite expensive screening for biomarker-positive patients. Available data support approval of new drugs without randomized trials if they yield single-agent sustained responses in patients refractory to standard therapies. Conversely, new approaches are needed to guide development of drug combinations since both standard phase II approaches and phase II-III randomized trials have a high risk of misleading.
Summary: Traditional randomized clinical trials approaches are often inefficient, wasteful, and unreliable. New clinical research paradigms are needed. The primary outcome of clinical research should be "Who (if anyone) benefits?" rather than "Does the overall group benefit?"
Figures
References
-
- Masia N, In: Focus on Intellectual Property Rights. edn. The cost of developing a new drug. Washington, D.C.: US Department of State Bureau of International Information Programs: Edited by Clack G, Neely MS; 2008. pp. 82–83.
-
- Silverman E. Clinical Trials Costs are Rising Rapidly. (posted 07/26/11): http://www.pharmalive.com/clinical-trial-costs-are-rising-rapidly.
-
- Patlak M, Nass S. Improving the quality of cancer clinical trials. National Academies Press: workshop summary; 2008.
-
- Stewart DJ, Whitney SN, Kurzrock R. Equipoise lost: ethics, costs, and the regulation of cancer clinical research. J Clin Oncol. 2010;28(17):2925–2935. - PubMed
-
- Arrondeau J, Gan HK, Razak AR, Paoletti X, Le Tourneau C. Development of anti-cancer drugs. Discovery Medicine. 2010;10(53):355–362. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
