

Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities
- PMID: 23587880
- PMCID: PMC4756476
- DOI: 10.1038/mp.2013.42
Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities
Abstract
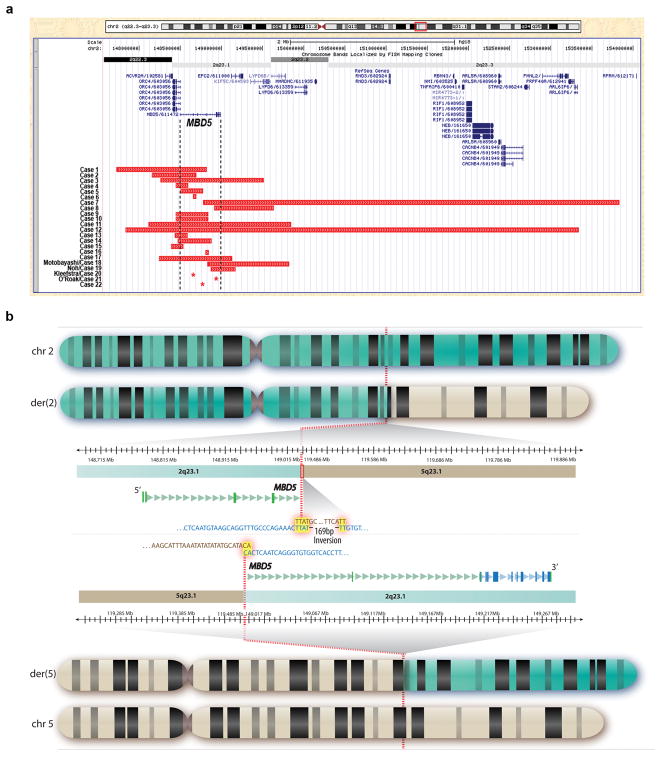
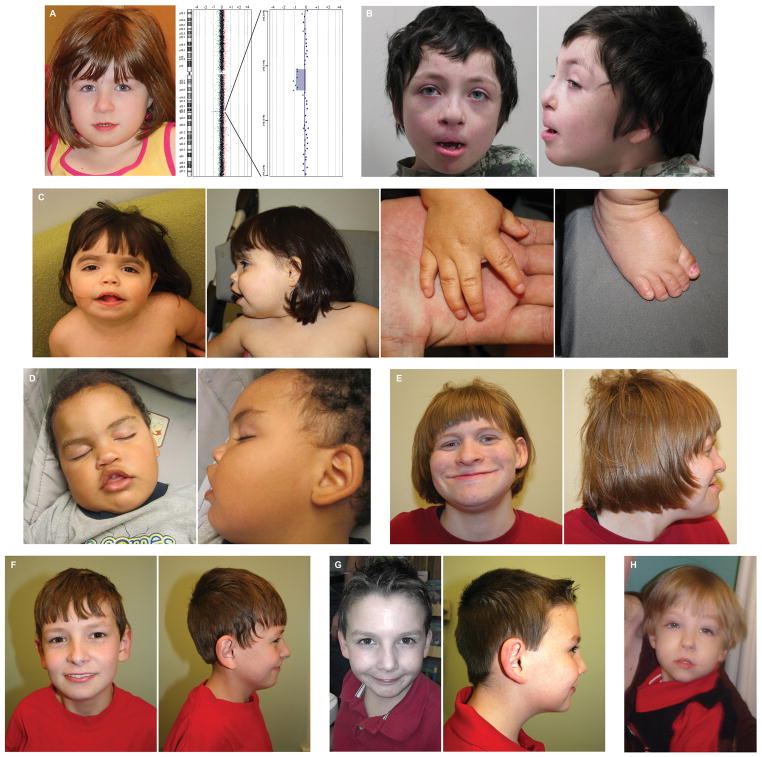
Microdeletions of chromosomal region 2q23.1 that disrupt MBD5 (methyl-CpG-binding domain protein 5) contribute to a spectrum of neurodevelopmental phenotypes; however, the impact of this locus on human psychopathology has not been fully explored. To characterize the structural variation landscape of MBD5 disruptions and the associated human psychopathology, 22 individuals with genomic disruption of MBD5 (translocation, point mutation and deletion) were identified through whole-genome sequencing or cytogenomic microarray at 11 molecular diagnostic centers. The genomic impact ranged from a single base pair to 5.4 Mb. Parents were available for 11 cases, all of which confirmed that the rearrangement arose de novo. Phenotypes were largely indistinguishable between patients with full-segment 2q23.1 deletions and those with intragenic MBD5 rearrangements, including alterations confined entirely to the 5'-untranslated region, confirming the critical impact of non-coding sequence at this locus. We identified heterogeneous, multisystem pathogenic effects of MBD5 disruption and characterized the associated spectrum of psychopathology, including the novel finding of anxiety and bipolar disorder in multiple patients. Importantly, one of the unique features of the oldest known patient was behavioral regression. Analyses also revealed phenotypes that distinguish MBD5 disruptions from seven well-established syndromes with significant diagnostic overlap. This study demonstrates that haploinsufficiency of MBD5 causes diverse phenotypes, yields insight into the spectrum of resulting neurodevelopmental and behavioral psychopathology and provides clinical context for interpretation of MBD5 structural variations. Empirical evidence also indicates that disruption of non-coding MBD5 regulatory regions is sufficient for clinical manifestation, highlighting the limitations of exon-focused assessments. These results suggest an ongoing perturbation of neurological function throughout the lifespan, including risks for neurobehavioral regression.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11. 2 and autism. N Engl J Med. 2008;358(7):667–675. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
