

Another fork in the road--life or death decisions by the tumour suppressor p53
- PMID: 23588418
- PMCID: PMC3642373
- DOI: 10.1038/embor.2013.25
Another fork in the road--life or death decisions by the tumour suppressor p53
Abstract
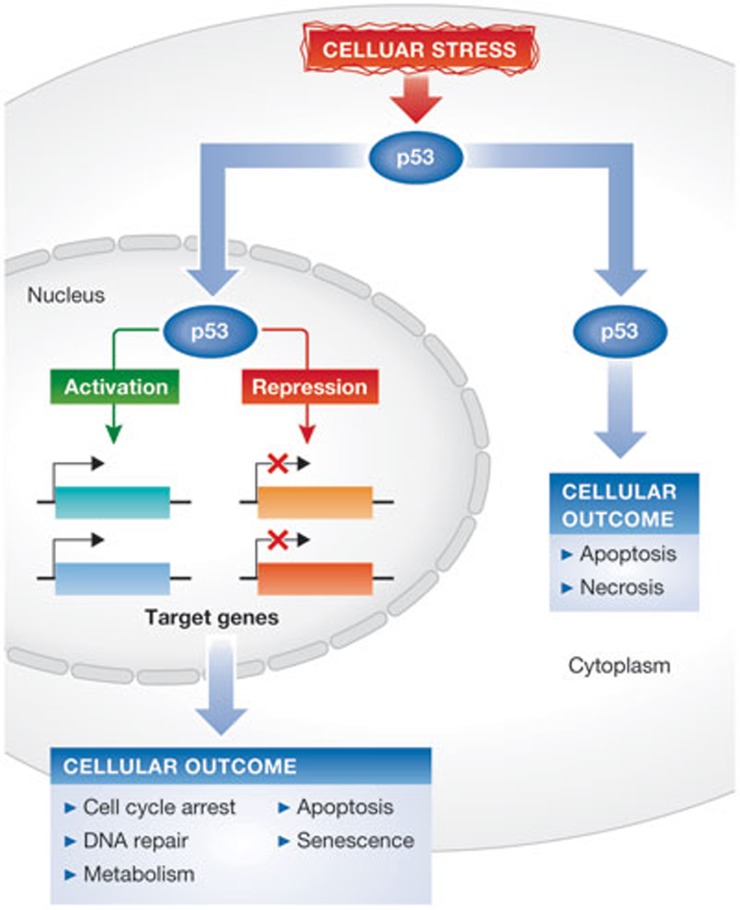
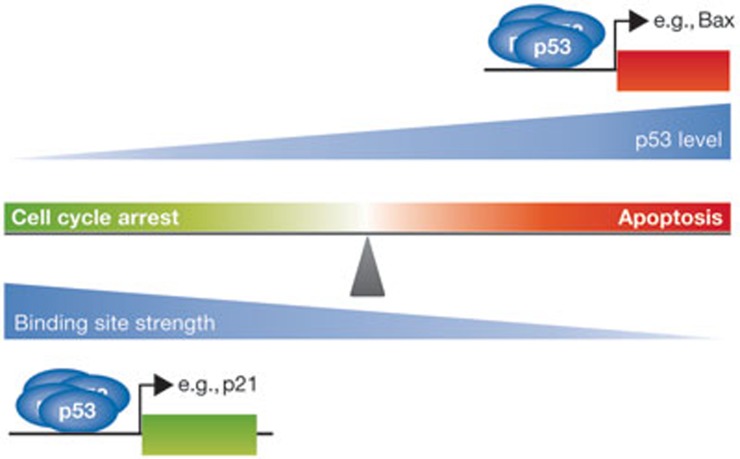
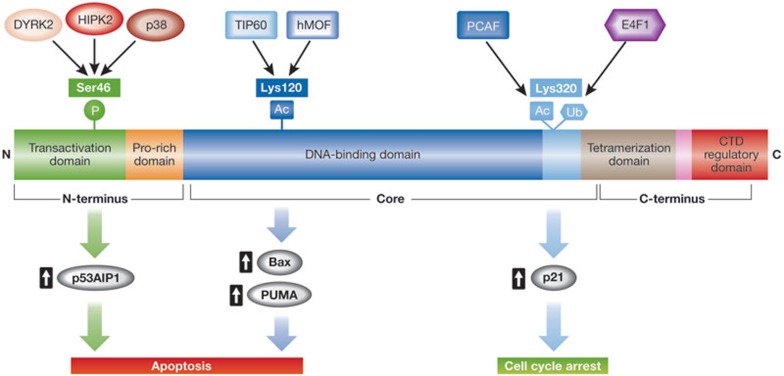
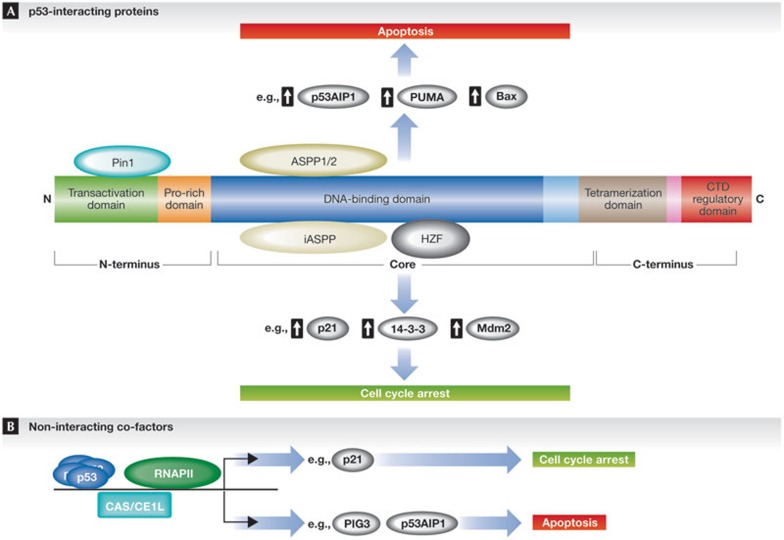
In response to cellular stress signals, the tumour suppressor p53 accumulates and triggers a host of antineoplastic responses. For instance, DNA damage activates two main p53-dependent responses: cell cycle arrest and attendant DNA repair or apoptosis (cell death). It is broadly accepted that, in response to DNA damage, the function of p53 as a sequence-specific transcription factor is crucial for tumour suppression. The molecular determinants, however, that favour the initiation of either a p53-dependent cell cycle arrest (life) or apoptotic (death) transcriptional programme remain elusive. Gaining a clear understanding of the mechanisms controlling cell fate determination by p53 could lead to the identification of molecular targets for therapy, which could selectively sensitize cancer cells to apoptosis. This review summarizes the literature addressing this important question in the field. Special emphasis is given to the role of the p53 response element, post-translational modifications and protein-protein interactions on cell fate decisions made by p53 in response to DNA damage.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 - PubMed
-
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 - PubMed
-
- Horn HF, Vousden KH (2007) Coping with stress: multiple ways to activate p53. Oncogene 26: 1306–1316 - PubMed
-
- Toledo F, Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 6: 909–923 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
