

Divergent mitochondrial biogenesis responses in human cardiomyopathy
- PMID: 23589024
- PMCID: PMC4236313
- DOI: 10.1161/CIRCULATIONAHA.112.001219
Divergent mitochondrial biogenesis responses in human cardiomyopathy
Abstract
Background: Mitochondria are key players in the development and progression of heart failure (HF). Mitochondrial (mt) dysfunction leads to diminished energy production and increased cell death contributing to the progression of left ventricular failure. The fundamental mechanisms that underlie mt dysfunction in HF have not been fully elucidated.
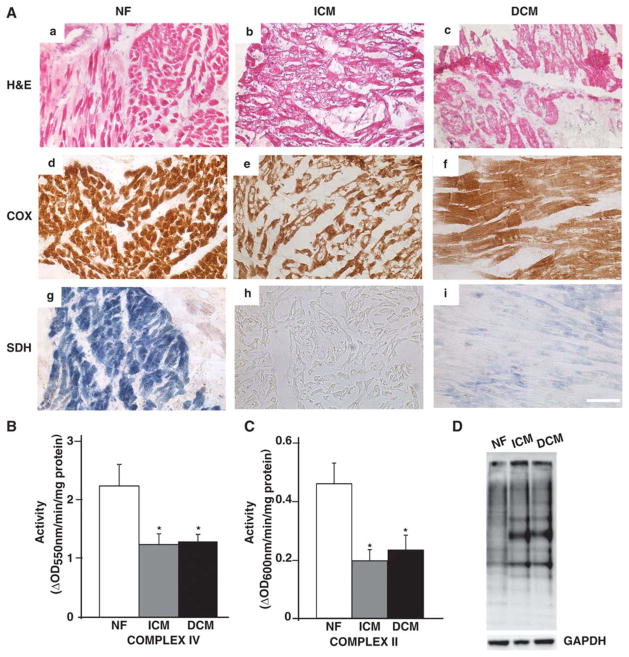
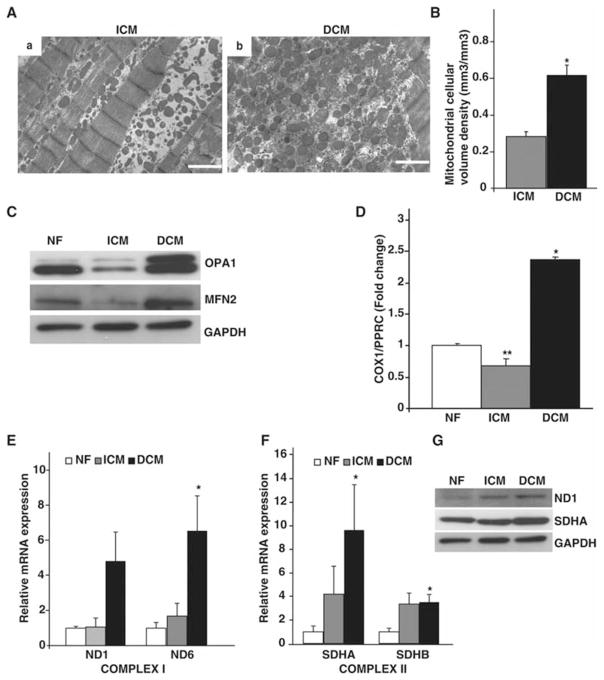
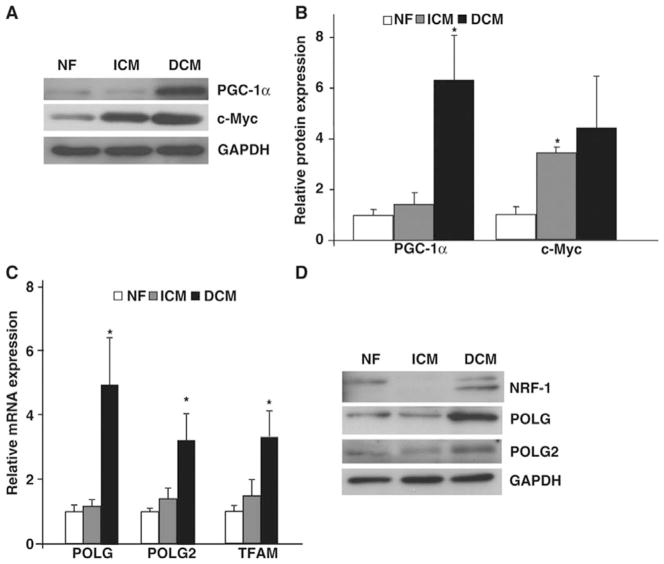
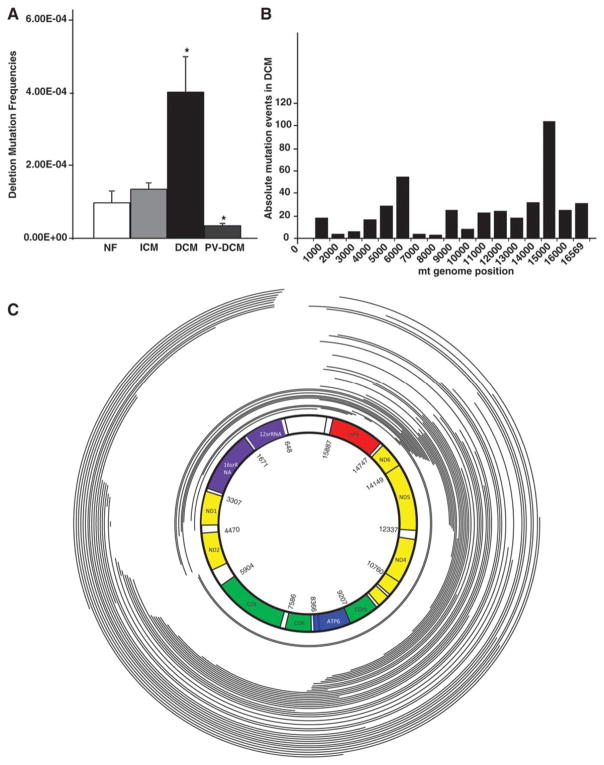
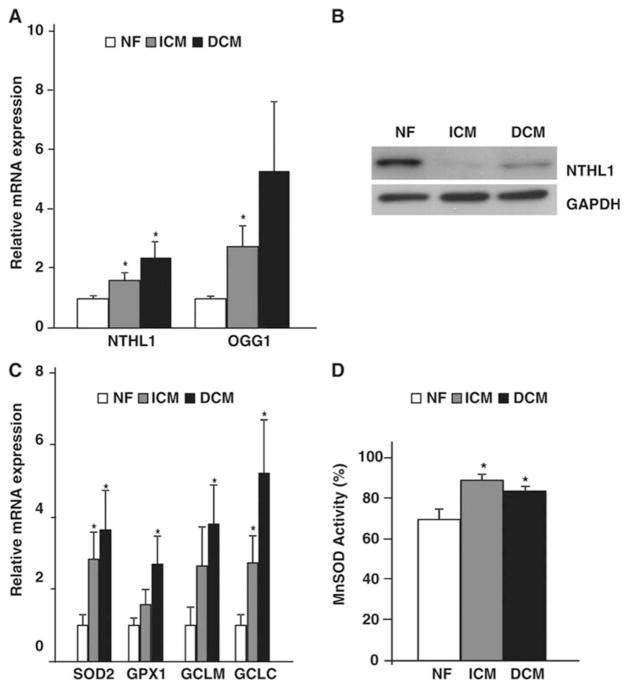
Methods and results: To characterize mt morphology, biogenesis, and genomic integrity in human HF, we investigated left ventricular tissue from nonfailing hearts and end-stage ischemic (ICM) or dilated (DCM) cardiomyopathic hearts. Although mt dysfunction was present in both types of cardiomyopathy, mt were smaller and increased in number in DCM compared with ICM or nonfailing hearts. mt volume density and mtDNA copy number was increased by ≈2-fold (P<0.001) in DCM hearts in comparison with ICM hearts. These changes were accompanied by an increase in the expression of mtDNA-encoded genes in DCM versus no change in ICM. mtDNA repair and antioxidant genes were reduced in failing hearts, suggestive of a defective repair and protection system, which may account for the 4.1-fold increase in mtDNA deletion mutations in DCM (P<0.05 versus nonfailing hearts, P<0.05 versus ICM).
Conclusions: In DCM, mt dysfunction is associated with mtDNA damage and deletions, which could be a consequence of mutating stress coupled with a peroxisome proliferator-activated receptor γ coactivator 1α-dependent stimulus for mt biogenesis. However, this maladaptive compensatory response contributes to additional oxidative damage. Thus, our findings support further investigations into novel mechanisms and therapeutic strategies for mt dysfunction in DCM.
Keywords: DNA, mitochondrial; cardiomyopathy, dilated; heart failure; mitochondrial turnover.
Conflict of interest statement
Figures
Comment in
-
Interplay of mitochondrial biogenesis and oxidative stress in heart failure.Circulation. 2013 May 14;127(19):1932-4. doi: 10.1161/CIRCULATIONAHA.113.003177. Epub 2013 Apr 15. Circulation. 2013. PMID: 23589023 No abstract available.
References
-
- Capetanaki Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc Med. 2002;12:339–348. - PubMed
-
- Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. - PubMed
-
- Marin-Garcia J, Goldenthal MJ. the mitochondrial organelle and the heart. Rev Esp Cardiol. 2002;55:1293–1310. - PubMed
-
- Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
