

Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer
- PMID: 23589849
- PMCID: PMC3645522
- DOI: 10.1073/pnas.1304977110
Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer
Abstract
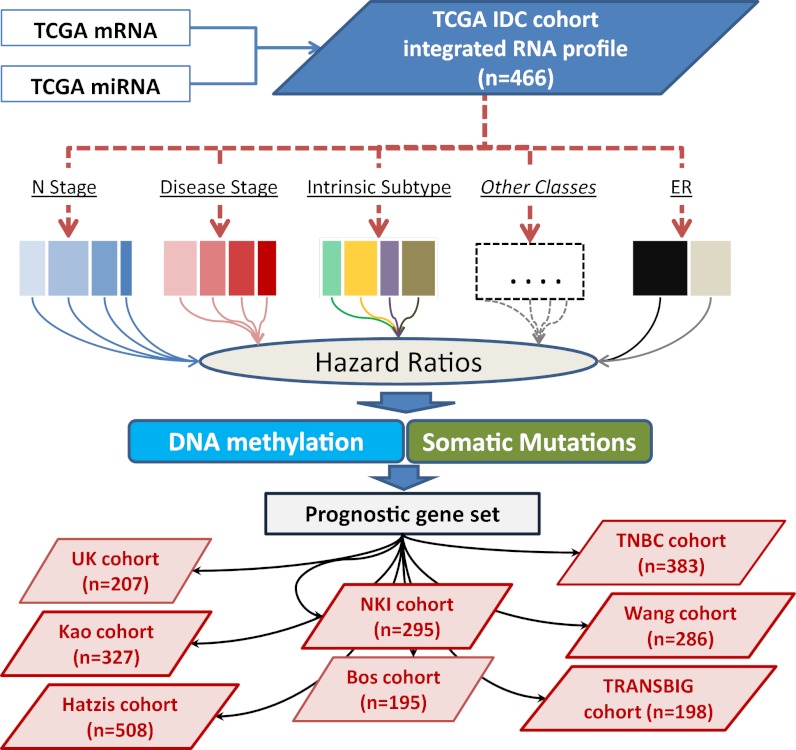
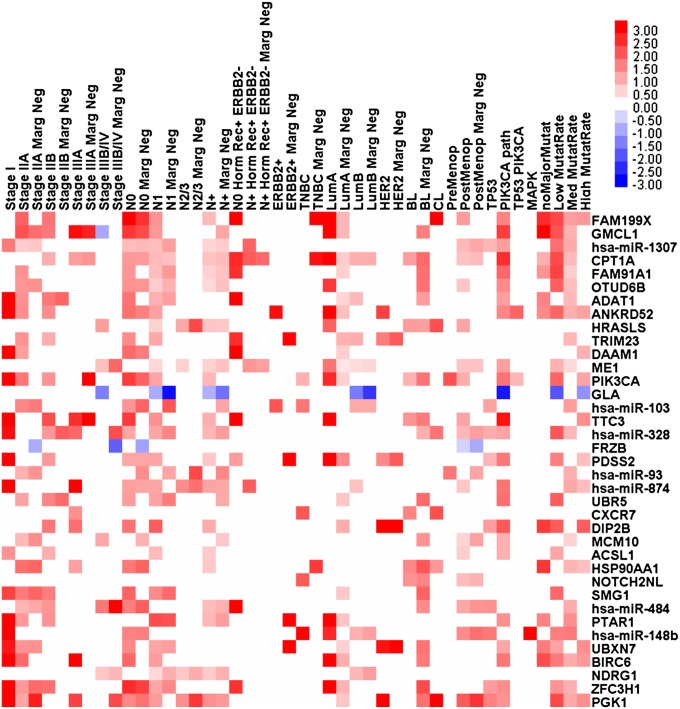
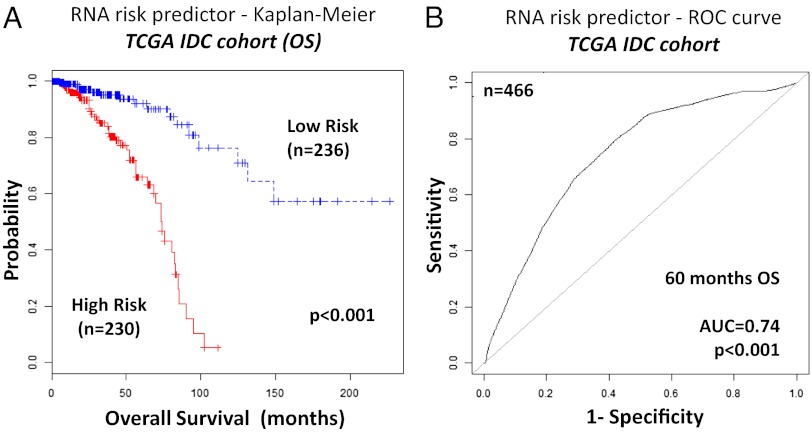
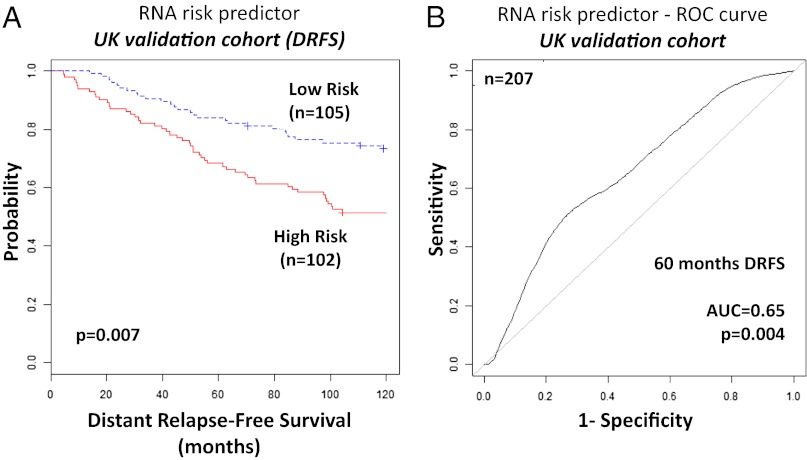
The optimal management of breast cancer (BC) presents challenges due to the heterogeneous molecular classification of the disease. We performed survival analysis on a cohort of 466 patients with primary invasive ductal carcinoma (IDC), the most frequent type of BC, by integrating mRNA, microRNA (miRNA), and DNA methylation next-generation sequencing data from The Cancer Genome Atlas (TCGA). Expression data from eight other BC cohorts were used for validation. The prognostic value of the resulting miRNA/mRNA signature was compared with that of other prognostic BC signatures. Thirty mRNAs and seven miRNAs were associated with overall survival across different clinical and molecular subclasses of a 466-patient IDC cohort from TCGA. The prognostic RNAs included PIK3CA, one of the two most frequently mutated genes in IDC, and miRNAs such as hsa-miR-328, hsa-miR-484, and hsa-miR-874. The area under the curve of the receiver-operator characteristic for the IDC risk predictor in the TCGA cohort was 0.74 at 60 mo of overall survival (P < 0.001). Most relevant for clinical application, the integrated signature had the highest prognostic value in early stage I and II tumors (receiver-operator characteristic area under the curve = 0.77, P value < 0.001). The genes in the RNA risk predictor had an independent prognostic value compared with the clinical covariates, as shown by multivariate analysis. The integrated RNA signature was successfully validated on eight BC cohorts, comprising a total of 2,399 patients, and it had superior performance for risk stratification with respect to other RNA predictors, including the mRNAs used in MammaPrint and Oncotype DX assays.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Shipitsin M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
