

Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation
- PMID: 23590265
- PMCID: PMC3734608
- DOI: 10.1164/rccm.201301-0153OC
Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation
Abstract
Rationale: Ivacaftor (VX-770), a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator, has been shown to improve lung function, pulmonary exacerbation rate, respiratory symptoms, and weight gain compared with placebo in patients with cystic fibrosis aged 12 years or older with a G551D-CFTR mutation.
Objectives: This randomized, double-blind, placebo-controlled trial evaluated ivacaftor in patients with cystic fibrosis aged 6-11 years with a G551D-CFTR mutation on at least one allele.
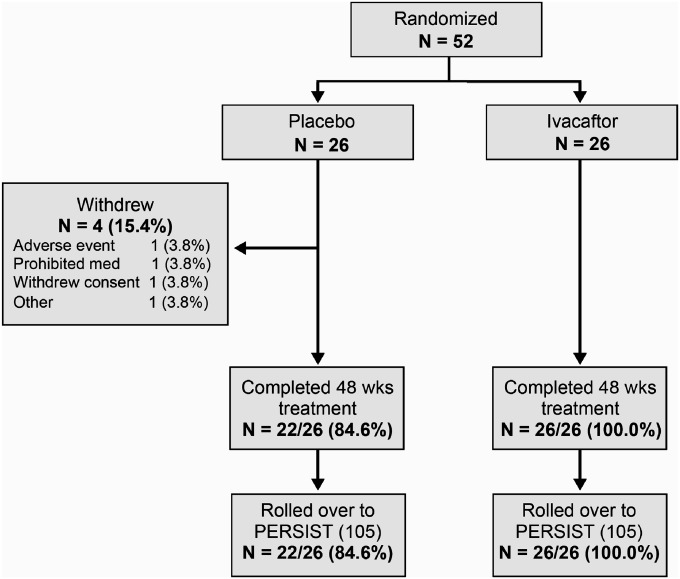
Methods: Patients were randomly assigned to receive ivacaftor administered orally at 150 mg (n = 26) or placebo (n = 26) every 12 hours for 48 weeks in addition to existing prescribed cystic fibrosis therapies.
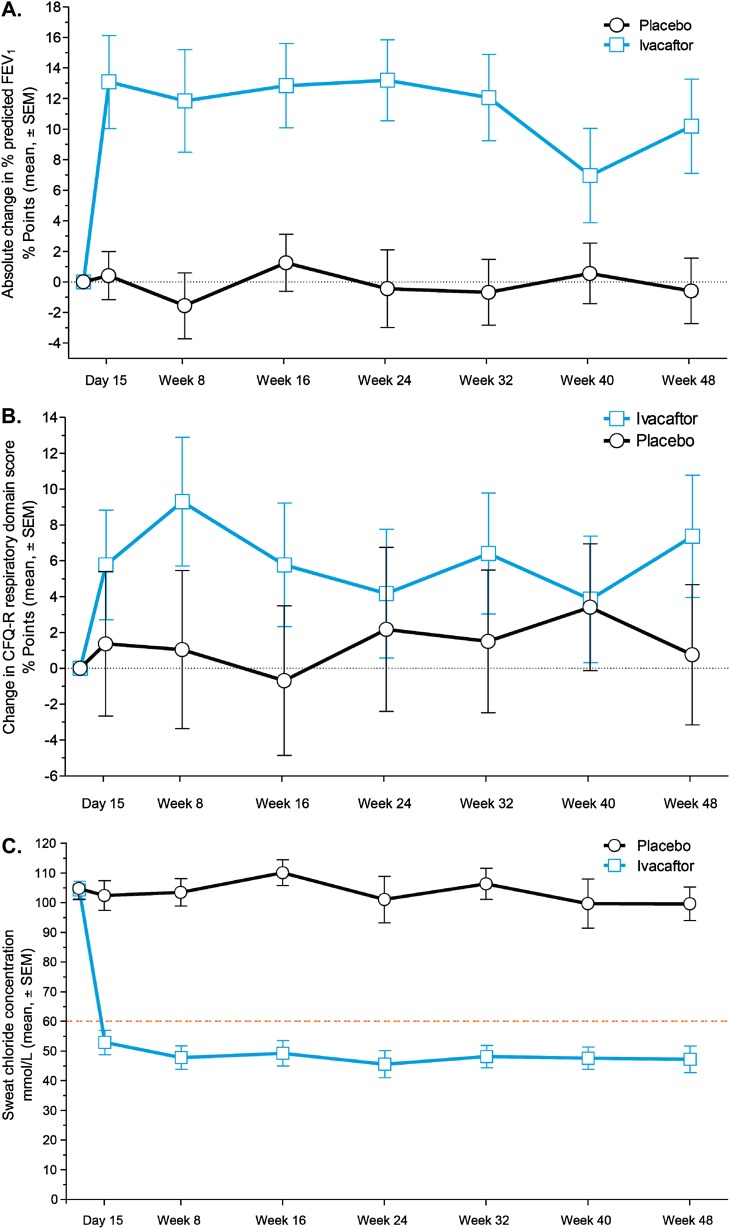
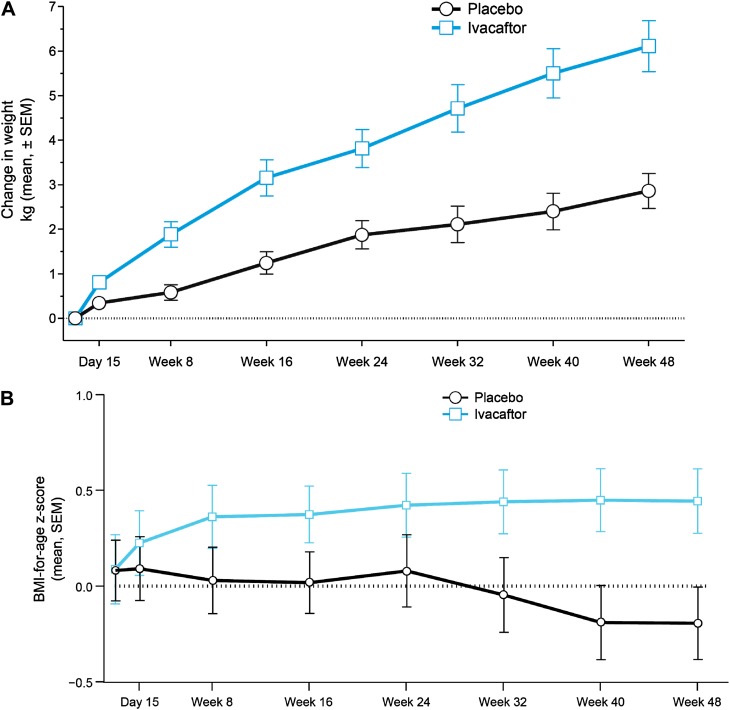
Measurements and main results: Despite near-normal mean baseline values in FEV1, patients receiving ivacaftor had a significant increase in percent predicted FEV1 from baseline through Week 24 versus placebo group (treatment effect, 12.5 percentage points; P < 0.001). Effects on pulmonary function were evident by 2 weeks, and a significant treatment effect was maintained through Week 48. Patients treated with ivacaftor gained, on average, 2.8 kg more than those receiving placebo at Week 48 (P < 0.001). The change from baseline through Week 48 in the concentration of sweat chloride, a measure of CFTR activity, with ivacaftor was -53.5 mmol/L (P < 0.001) versus placebo. The incidence of adverse events was similar in the two groups.
Conclusions: In patients who are younger and healthier than those in previously studied populations, ivacaftor demonstrated a significant improvement in pulmonary function, weight, and CFTR activity compared with placebo. Clinical trial registered with www.clinicaltrials.gov (NCT00909727).
Figures
References
-
- Cystic Fibrosis Foundation. Bethesda, MD: Cystic Fibrosis Foundation; 2011. Cystic fibrosis foundation patient registry: 2010 annual data report.
-
- Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7:450–453. - PubMed
-
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
