

Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis
- PMID: 23592792
- PMCID: PMC3668729
- DOI: 10.1074/jbc.M112.425066
Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis
Abstract
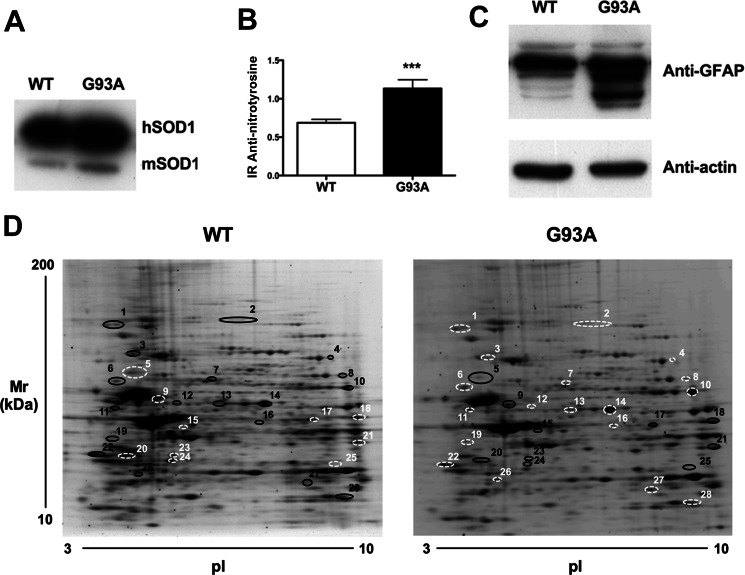
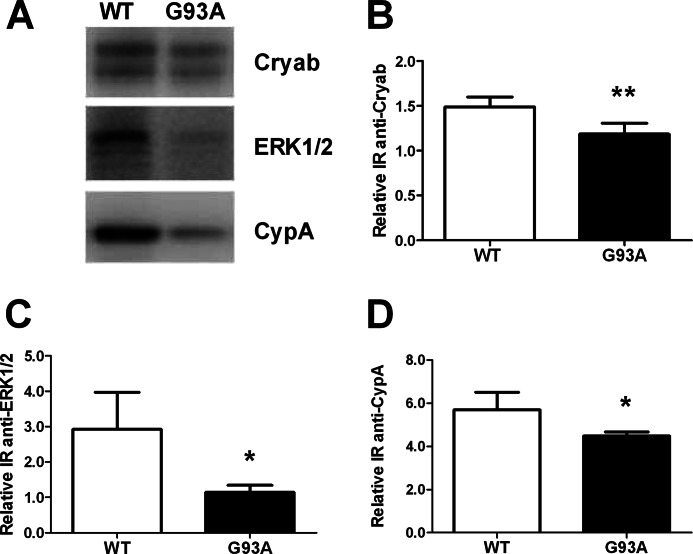
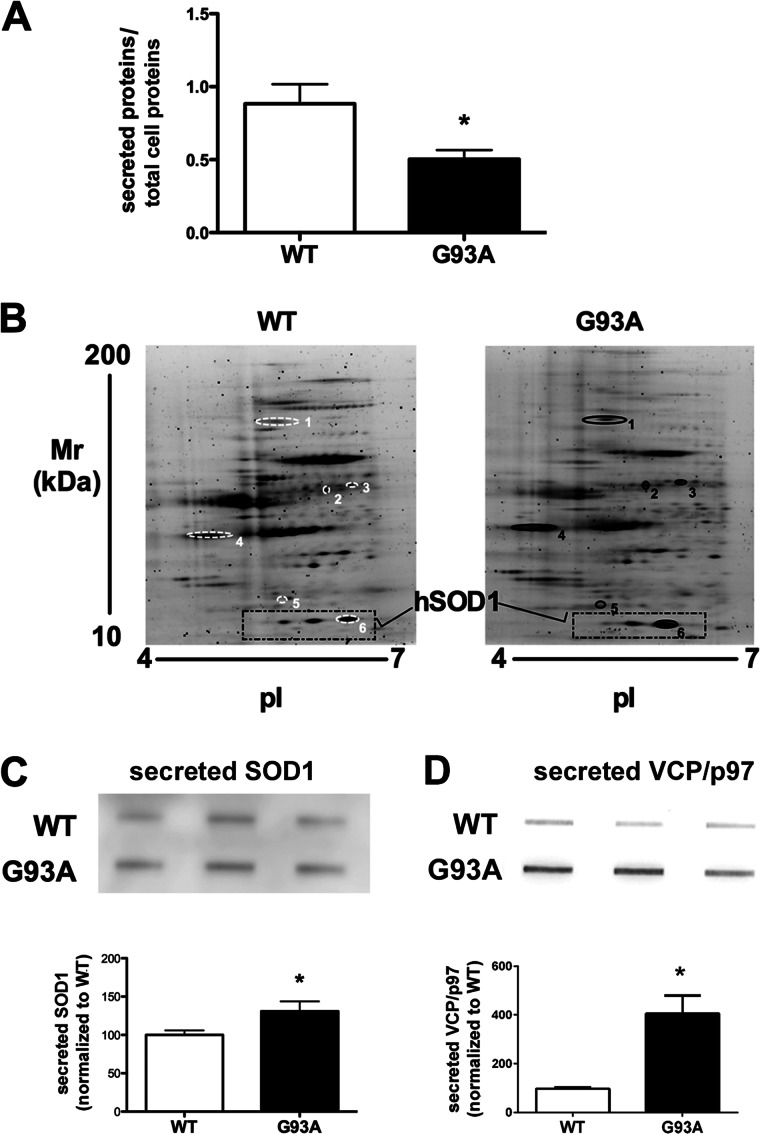
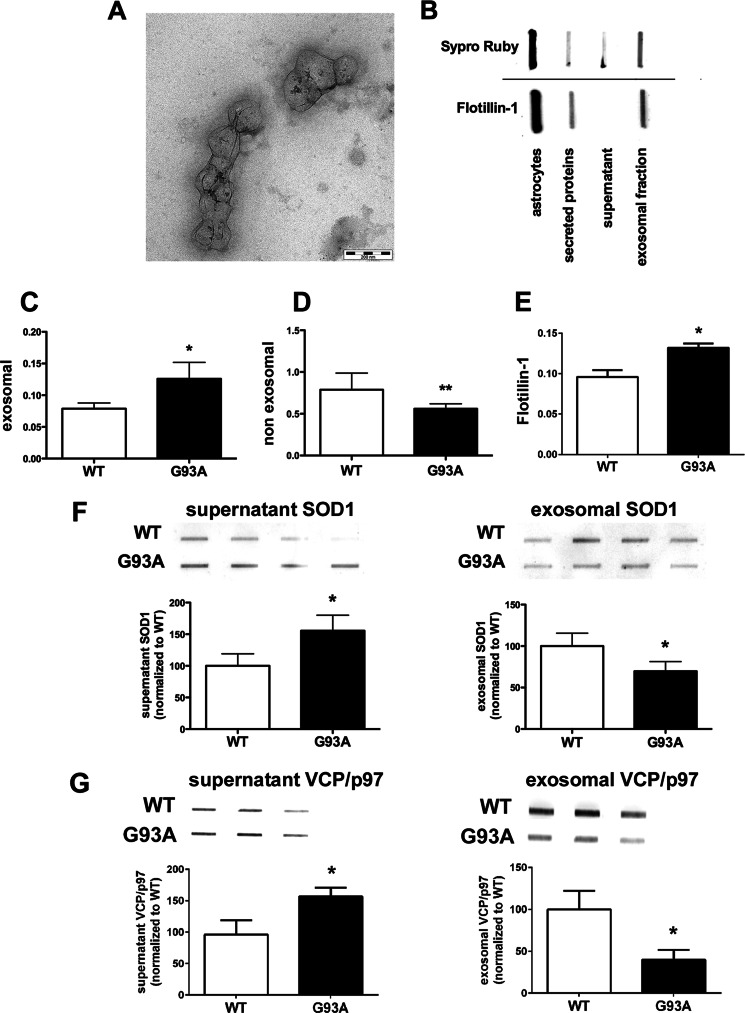
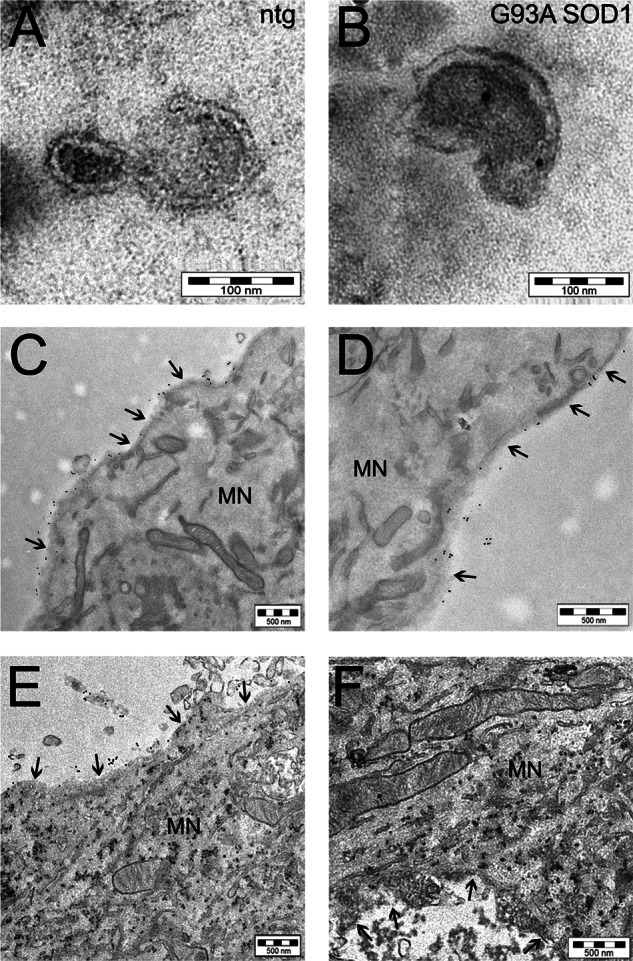
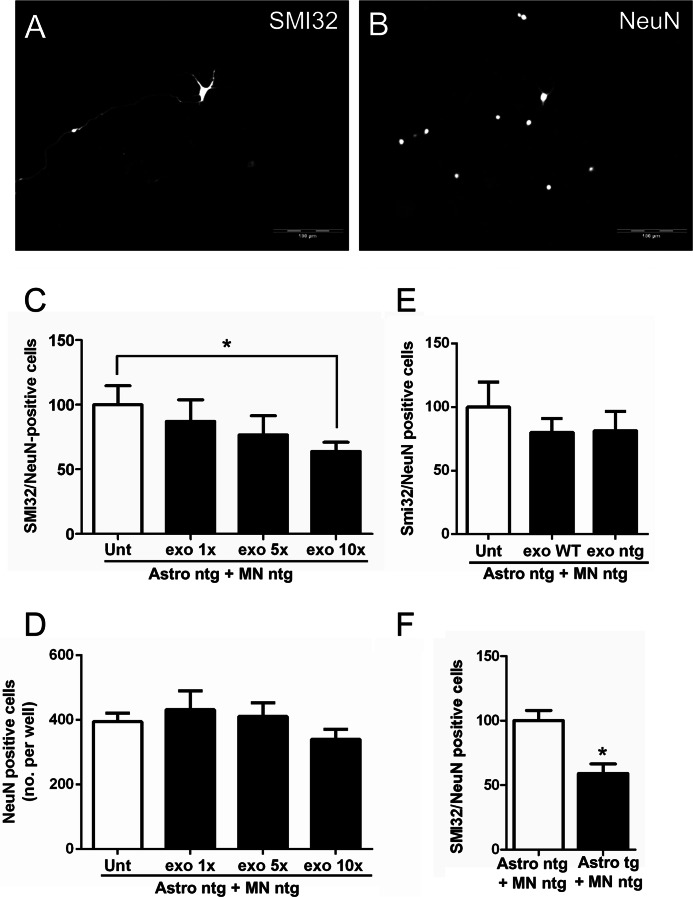
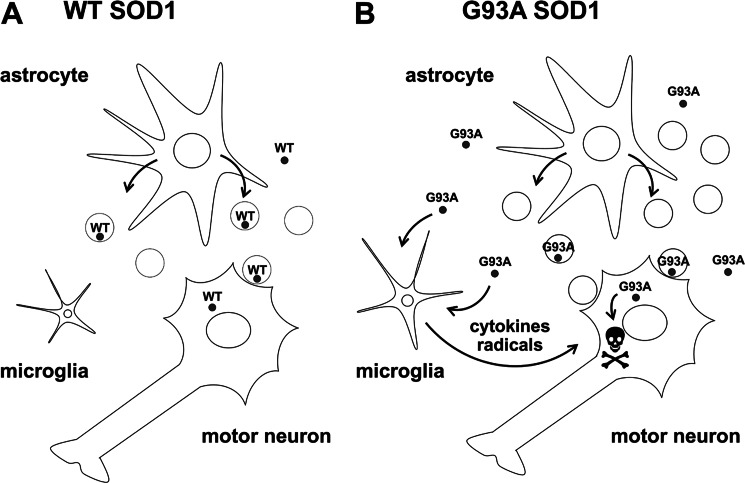
Amyotrophic lateral sclerosis is the most common motor neuron disease and is still incurable. The mechanisms leading to the selective motor neuron vulnerability are still not known. The interplay between motor neurons and astrocytes is crucial in the outcome of the disease. We show that mutant copper-zinc superoxide dismutase (SOD1) overexpression in primary astrocyte cultures is associated with decreased levels of proteins involved in secretory pathways. This is linked to a general reduction of total secreted proteins, except for specific enrichment in a number of proteins in the media, such as mutant SOD1 and valosin-containing protein (VCP)/p97. Because there was also an increase in exosome release, we can deduce that astrocytes expressing mutant SOD1 activate unconventional secretory pathways, possibly as a protective mechanism. This may help limit the formation of intracellular aggregates and overcome mutant SOD1 toxicity. We also found that astrocyte-derived exosomes efficiently transfer mutant SOD1 to spinal neurons and induce selective motor neuron death. We conclude that the expression of mutant SOD1 has a substantial impact on astrocyte protein secretion pathways, contributing to motor neuron pathology and disease spread.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig's Disease); Astrocytes; Disease Spreading; Exosomes; Proteomics; Superoxide Dismutase (SOD).
Figures
References
-
- Howland D. S., Liu J., She Y., Goad B., Maragakis N. J., Kim B., Erickson J., Kulik J., DeVito L., Psaltis G., DeGennaro L. J., Cleveland D. W., Rothstein J. D. (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. U.S.A. 99, 1604–1609 - PMC - PubMed
-
- Rothstein J. D., Van Kammen M., Levey A. I., Martin L. J., Kuncl R. W. (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84 - PubMed
-
- Bendotti C., Tortarolo M., Suchak S. K., Calvaresi N., Carvelli L., Bastone A., Rizzi M., Rattray M., Mennini T. (2001) Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J. Neurochem. 79, 737–746 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
