

Comparative genomics of Wolbachia and the bacterial species concept
- PMID: 23593012
- PMCID: PMC3616963
- DOI: 10.1371/journal.pgen.1003381
Comparative genomics of Wolbachia and the bacterial species concept
Abstract
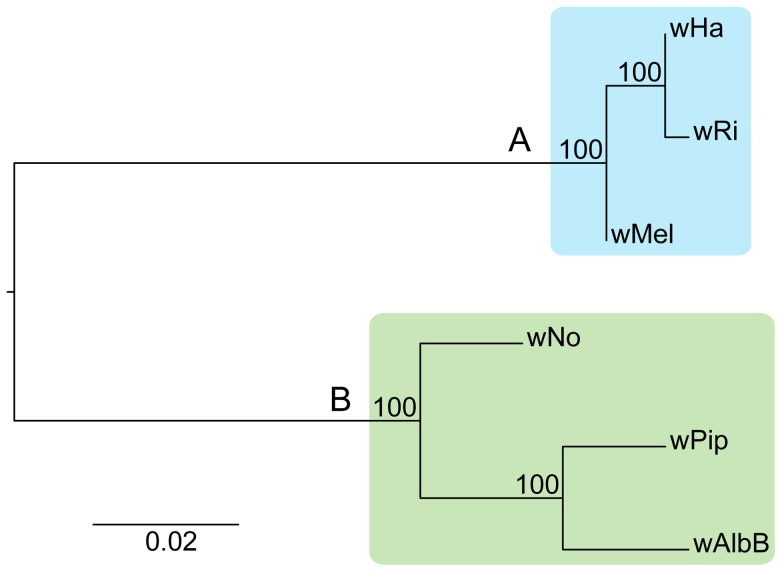
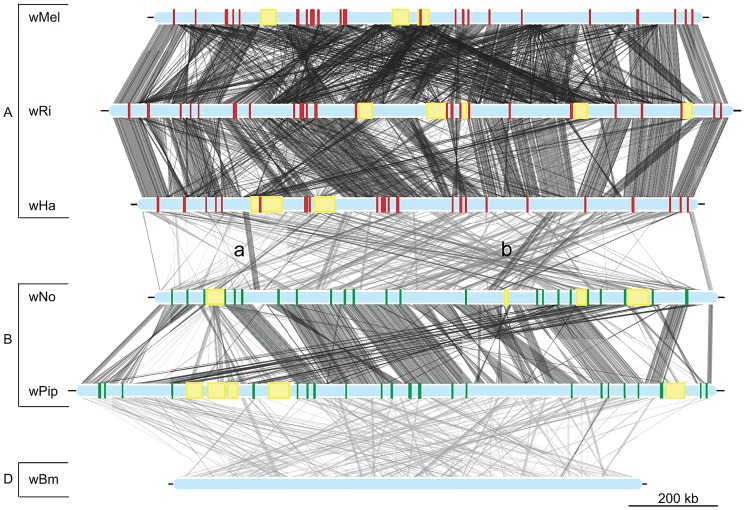
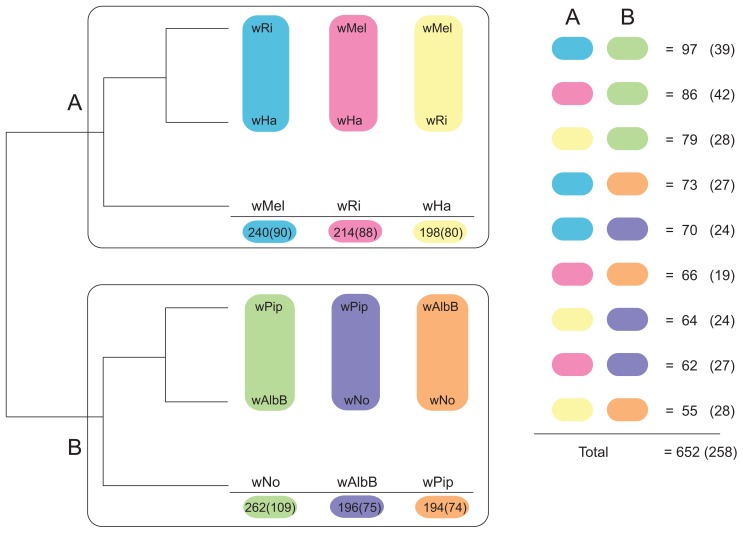
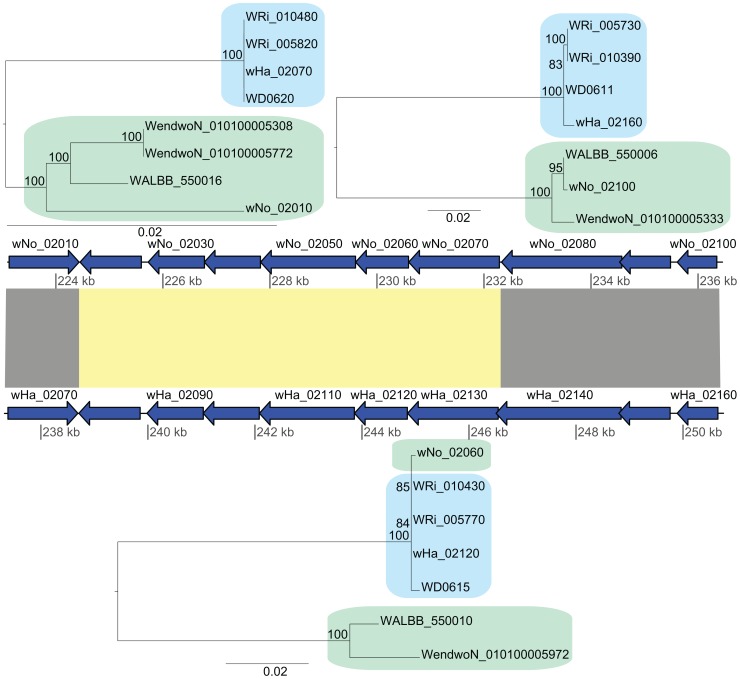
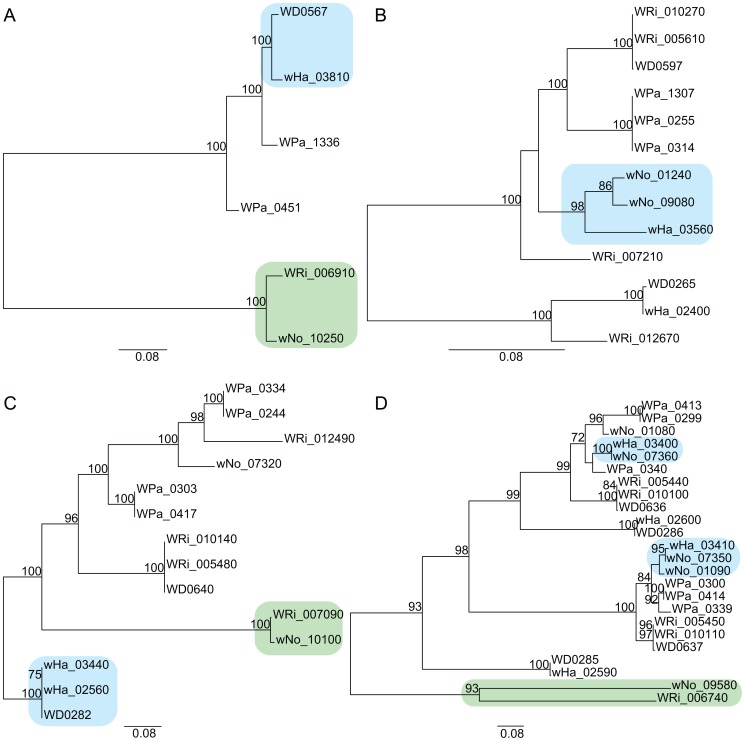
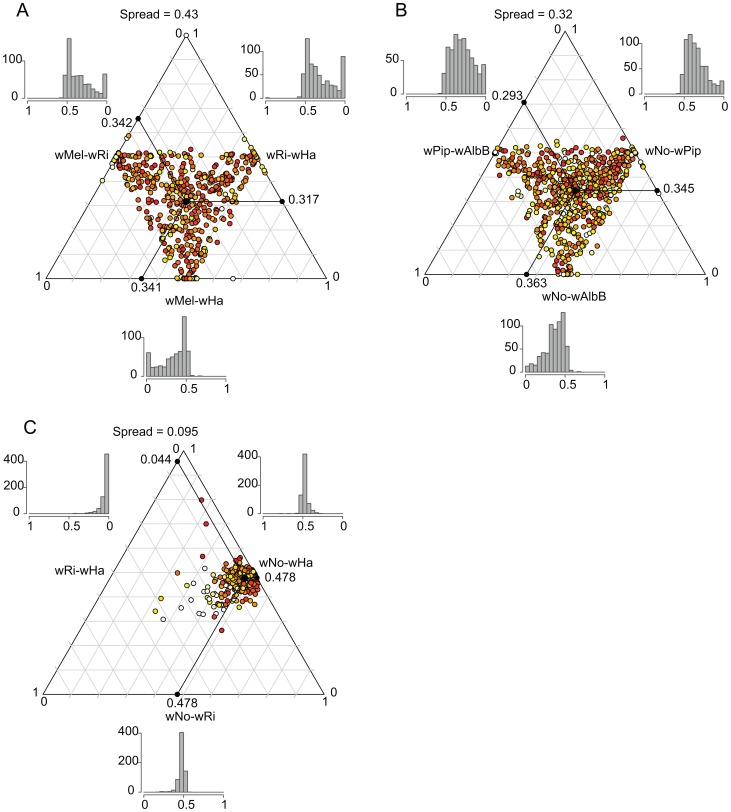
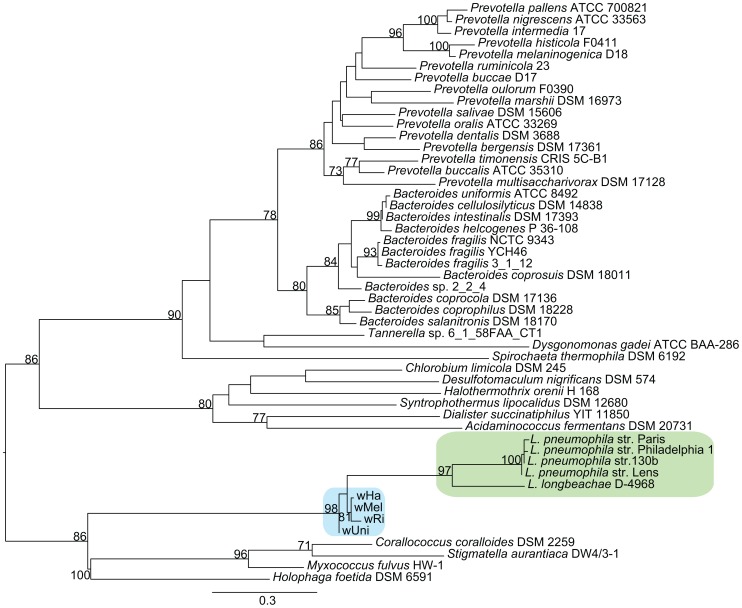
The importance of host-specialization to speciation processes in obligate host-associated bacteria is well known, as is also the ability of recombination to generate cohesion in bacterial populations. However, whether divergent strains of highly recombining intracellular bacteria, such as Wolbachia, can maintain their genetic distinctness when infecting the same host is not known. We first developed a protocol for the genome sequencing of uncultivable endosymbionts. Using this method, we have sequenced the complete genomes of the Wolbachia strains wHa and wNo, which occur as natural double infections in Drosophila simulans populations on the Seychelles and in New Caledonia. Taxonomically, wHa belong to supergroup A and wNo to supergroup B. A comparative genomics study including additional strains supported the supergroup classification scheme and revealed 24 and 33 group-specific genes, putatively involved in host-adaptation processes. Recombination frequencies were high for strains of the same supergroup despite different host-preference patterns, leading to genomic cohesion. The inferred recombination fragments for strains of different supergroups were of short sizes, and the genomes of the co-infecting Wolbachia strains wHa and wNo were not more similar to each other and did not share more genes than other A- and B-group strains that infect different hosts. We conclude that Wolbachia strains of supergroup A and B represent genetically distinct clades, and that strains of different supergroups can co-exist in the same arthropod host without converging into the same species. This suggests that the supergroups are irreversibly separated and that barriers other than host-specialization are able to maintain distinct clades in recombining endosymbiont populations. Acquiring a good knowledge of the barriers to genetic exchange in Wolbachia will advance our understanding of how endosymbiont communities are constructed from vertically and horizontally transmitted genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Diminutive, degraded but dissimilar: Wolbachia genomes from filarial nematodes do not conform to a single paradigm.Microb Genom. 2020 Dec;6(12):mgen000487. doi: 10.1099/mgen.0.000487. Epub 2020 Dec 9. Microb Genom. 2020. PMID: 33295865 Free PMC article.
-
Evolution of Wolbachia-induced cytoplasmic incompatibility in Drosophila simulans and D. sechellia.Evolution. 2002 Sep;56(9):1735-42. doi: 10.1111/j.0014-3820.2002.tb00187.x. Evolution. 2002. PMID: 12389718
-
Complete de novo assembly of Wolbachia endosymbiont of Drosophila willistoni using long-read genome sequencing.Sci Rep. 2024 Aug 1;14(1):17770. doi: 10.1038/s41598-024-68716-w. Sci Rep. 2024. PMID: 39090271 Free PMC article.
-
Wolbachia genomes: revealing the biology of parasitism and mutualism.Trends Parasitol. 2006 Feb;22(2):60-5. doi: 10.1016/j.pt.2005.12.012. Epub 2006 Jan 10. Trends Parasitol. 2006. PMID: 16406333 Review.
-
Wolbachia genomes: insights into an intracellular lifestyle.Curr Biol. 2005 Jul 12;15(13):R507-9. doi: 10.1016/j.cub.2005.06.029. Curr Biol. 2005. PMID: 16005284 Review.
Cited by
-
Evidence for common horizontal transmission of Wolbachia among butterflies and moths.BMC Evol Biol. 2016 May 27;16(1):118. doi: 10.1186/s12862-016-0660-x. BMC Evol Biol. 2016. PMID: 27233666 Free PMC article.
-
Two sympatric lineages of Australian Cnestus solidus share Ambrosiella symbionts but not Wolbachia.Heredity (Edinb). 2024 Jan;132(1):43-53. doi: 10.1038/s41437-023-00659-w. Epub 2023 Nov 10. Heredity (Edinb). 2024. PMID: 37949964 Free PMC article.
-
Evolution and population genomics of the Lyme borreliosis pathogen, Borrelia burgdorferi.Trends Genet. 2015 Apr;31(4):201-7. doi: 10.1016/j.tig.2015.02.006. Epub 2015 Mar 9. Trends Genet. 2015. PMID: 25765920 Free PMC article. Review.
-
Wolbachia Horizontal Transmission Events in Ants: What Do We Know and What Can We Learn?Front Microbiol. 2019 Mar 6;10:296. doi: 10.3389/fmicb.2019.00296. eCollection 2019. Front Microbiol. 2019. PMID: 30894837 Free PMC article.
-
Spatial and temporal sex ratio bias and Wolbachia-infection in New Zealand Crambidae (Lepidoptera: Pyraloidea).Biodivers Data J. 2020 Jul 7;8:e52621. doi: 10.3897/BDJ.8.e52621. eCollection 2020. Biodivers Data J. 2020. PMID: 32733140 Free PMC article.
References
-
- Mira A, Martin-Cuadrado AB, D'Auria G, Rodriguez-Valera F (2010) The bacterial pan-genome:a new paradigm in microbiology. Int Microbiol 13: 45–57. - PubMed
-
- Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, et al. (2005) Opinion: Re-evaluating prokaryotic species. Nat Rev Microbiol 3: 733–739. - PubMed
-
- Achtman M, Wagner M (2008) Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6: 431–440. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
