

Revising a personal genome by comparing and combining data from two different sequencing platforms
- PMID: 23593254
- PMCID: PMC3620462
- DOI: 10.1371/journal.pone.0060585
Revising a personal genome by comparing and combining data from two different sequencing platforms
Abstract
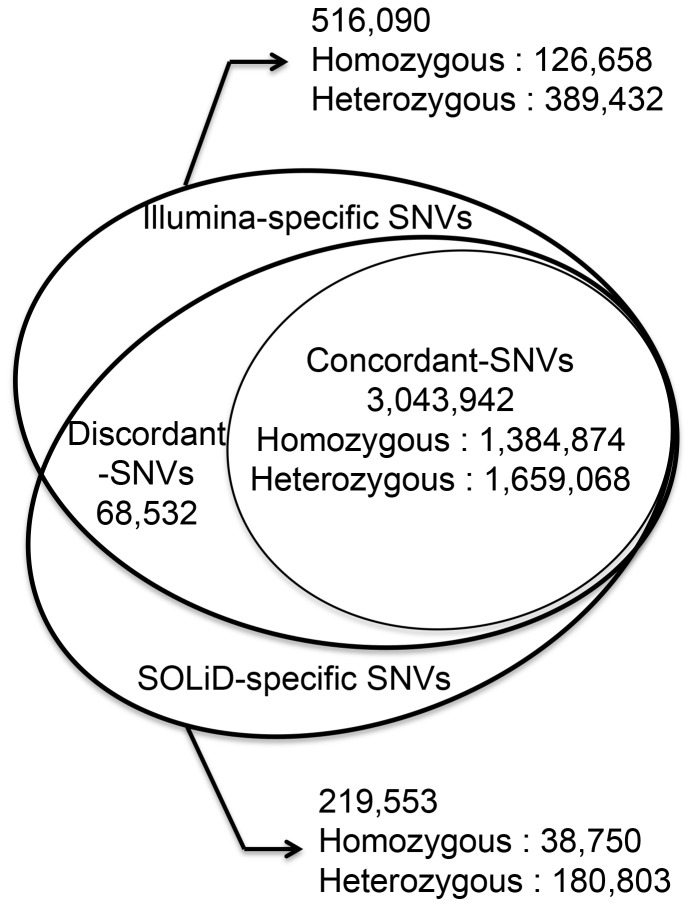
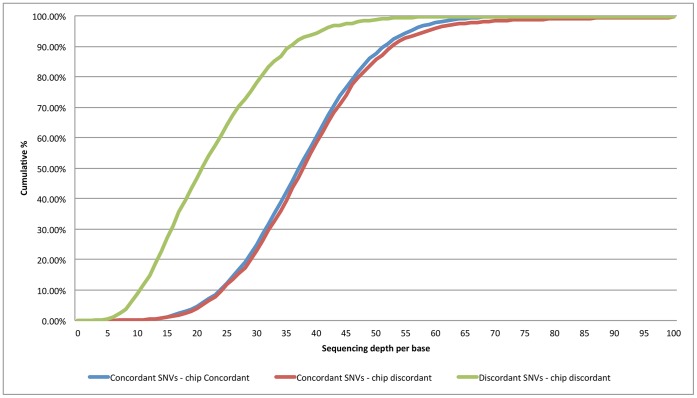
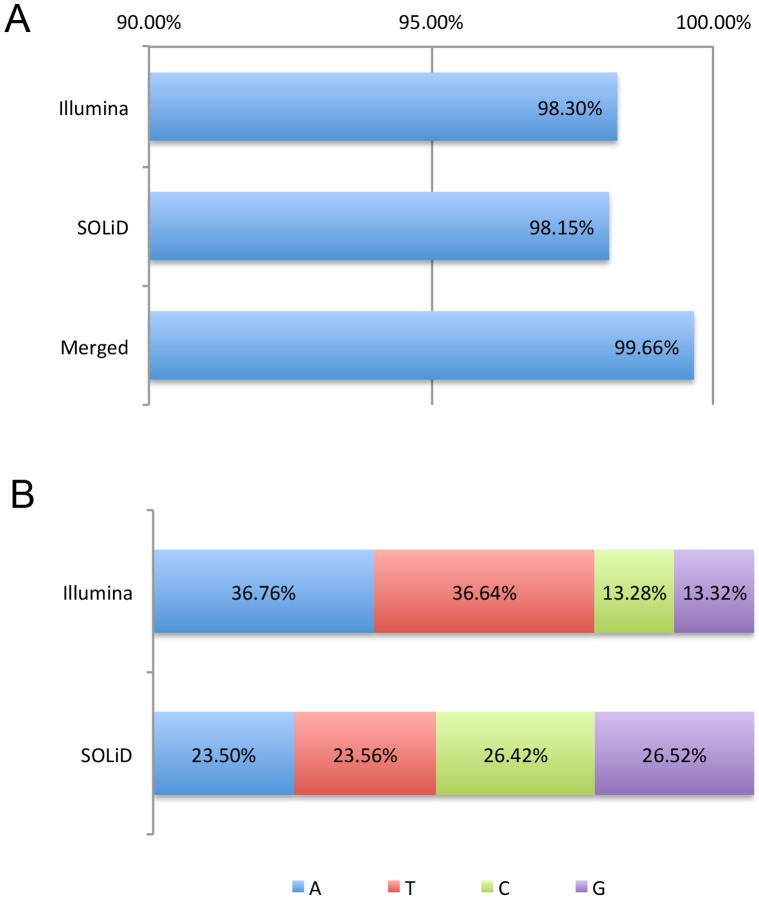
For the robust practice of genomic medicine, sequencing results must be compatible, regardless of the sequencing technologies and algorithms used. Presently, genome sequencing is still an imprecise science and is complicated by differences in the chemistry, coverage, alignment, and variant-calling algorithms. We identified ~3.33 million single nucleotide variants (SNVs) and ~3.62 million SNVs in the SJK genome using SOLiD and Illumina data, respectively. Approximately 3 million SNVs were concordant between the two platforms while 68,532 SNVs were discordant; 219,616 SNVs were SOLiD-specific and 516,080 SNVs were Illumina-specific (i.e., platform-specific). Concordant, discordant, and platform-specific SNVs were further analyzed and characterized. Overall, a large portion of heterozygous SNVs that were discordant with genotyping calls of single nucleotide polymorphism chips were highly confident. Approximately 70% of the platform-specific SNVs were located in regions containing repetitive sequences. Such platform-specificity may arise from differences between platforms, with regard to read length (36 bp and 72 bp vs. 50 bp), insert size (~100-300 bp vs. ~1-2 kb), sequencing chemistry (sequencing-by-synthesis using single nucleotides vs. ligation-based sequencing using oligomers), and sequencing quality. When data from the two platforms were merged for variant calling, the proportion of callable regions of the reference genome increased to 99.66%, which was 1.43% higher than the average callability of the two platforms, representing ~40 million bases. In this study, we compared the differences in sequencing results between two sequencing platforms. Approximately 90% of the SNVs were concordant between the two platforms, yet ~10% of the SNVs were either discordant or platform-specific, indicating that each platform had its own strengths and weaknesses. When data from the two platforms were merged, both the overall callability of the reference genome and the overall accuracy of the SNVs improved, demonstrating the likelihood that a re-sequenced genome can be revised using complementary data.
Conflict of interest statement
Figures
Similar articles
-
Performance comparison of whole-genome sequencing platforms.Nat Biotechnol. 2011 Dec 18;30(1):78-82. doi: 10.1038/nbt.2065. Nat Biotechnol. 2011. PMID: 22178993 Free PMC article.
-
Short and long-read genome sequencing methodologies for somatic variant detection; genomic analysis of a patient with diffuse large B-cell lymphoma.Sci Rep. 2021 Mar 19;11(1):6408. doi: 10.1038/s41598-021-85354-8. Sci Rep. 2021. PMID: 33742045 Free PMC article.
-
Coverage bias and sensitivity of variant calling for four whole-genome sequencing technologies.PLoS One. 2013 Jun 11;8(6):e66621. doi: 10.1371/journal.pone.0066621. Print 2013. PLoS One. 2013. PMID: 23776689 Free PMC article.
-
Towards population-scale long-read sequencing.Nat Rev Genet. 2021 Sep;22(9):572-587. doi: 10.1038/s41576-021-00367-3. Epub 2021 May 28. Nat Rev Genet. 2021. PMID: 34050336 Free PMC article. Review.
-
Functional mapping of the human genome by cDNA localization versus sequencing.Bioessays. 1994 Sep;16(9):693-8. doi: 10.1002/bies.950160917. Bioessays. 1994. PMID: 7980497 Review. No abstract available.
Cited by
-
THE BENEFITS OF CUSTOMIZED DNA DIRECTED NUTRITION TO BALANCE THE BRAIN REWARD CIRCUITRY AND REDUCE ADDICTIVE BEHAVIORS.Precis Med (Bangalore). 2016;1(1):18-33. Epub 2016 Aug 1. Precis Med (Bangalore). 2016. PMID: 28066828 Free PMC article.
-
An emerging place for lung cancer genomics in 2013.J Thorac Dis. 2013 Oct;5 Suppl 5(Suppl 5):S491-7. doi: 10.3978/j.issn.2072-1439.2013.10.06. J Thorac Dis. 2013. PMID: 24163742 Free PMC article. Review.
-
Characterization of H5N1 Influenza Virus Quasispecies with Adaptive Hemagglutinin Mutations from Single-Virus Infections of Human Airway Cells.J Virol. 2018 May 14;92(11):e02004-17. doi: 10.1128/JVI.02004-17. Print 2018 Jun 1. J Virol. 2018. PMID: 29563293 Free PMC article.
References
-
- Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133–141. - PubMed
-
- Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26: 1135–1145. - PubMed
-
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, et al. (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
