

Sequence analysis of 96 genomic regions identifies distinct evolutionary lineages within CC156, the largest Streptococcus pneumoniae clonal complex in the MLST database
- PMID: 23593373
- PMCID: PMC3625235
- DOI: 10.1371/journal.pone.0061003
Sequence analysis of 96 genomic regions identifies distinct evolutionary lineages within CC156, the largest Streptococcus pneumoniae clonal complex in the MLST database
Abstract
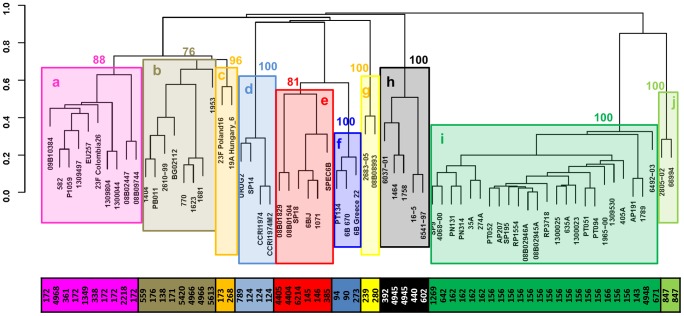
Multi-Locus Sequence Typing (MLST) of Streptococcus pneumoniae is based on the sequence of seven housekeeping gene fragments. The analysis of MLST allelic profiles by eBURST allows the grouping of genetically related strains into Clonal Complexes (CCs) including those genotypes with a common descent from a predicted ancestor. However, the increasing use of MLST to characterize S. pneumoniae strains has led to the identification of a large number of new Sequence Types (STs) causing the merger of formerly distinct lineages into larger CCs. An example of this is the CC156, displaying a high level of complexity and including strains with allelic profiles differing in all seven of the MLST loci, capsular type and the presence of the Pilus Islet-1 (PI-1). Detailed analysis of the CC156 indicates that the identification of new STs, such as ST4945, induced the merging of formerly distinct clonal complexes. In order to discriminate the strain diversity within CC156, a recently developed typing schema, 96-MLST, was used to analyse 66 strains representative of 41 different STs. Analysis of allelic profiles by hierarchical clustering and a minimum spanning tree identified ten genetically distinct evolutionary lineages. Similar results were obtained by phylogenetic analysis on the concatenated sequences with different methods. The identified lineages are homogenous in capsular type and PI-1 presence. ST4945 strains were unequivocally assigned to one of the lineages. In conclusion, the identification of new STs through an exhaustive analysis of pneumococcal strains from various laboratories has highlighted that potentially unrelated subgroups can be grouped into a single CC by eBURST. The analysis of additional loci, such as those included in the 96-MLST schema, will be necessary to accurately discriminate the clonal evolution of the pneumococcal population.
Conflict of interest statement
Figures




References
-
- O’Brien KL, Wolfson LJ, Watt JP, Henkle E, oria-Knoll M, et al. (2009) Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374: 893–902. - PubMed
-
- van der Poll T, Opal SM (2009) Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374: 1543–1556. - PubMed
-
- Weiser JN (2010) The pneumococcus: why a commensal misbehaves. J Mol Med 88: 97–102 doi:10.1007/s00109-009-0557-x. - DOI - PMC - PubMed
-
- Pelton SI, Leibovitz E (2009) Recent advances in otitis media. Pediatr Infect Dis J 28: S133–S137. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
