

Phylogenetic and structural diversity in the feline leukemia virus env gene
- PMID: 23593376
- PMCID: PMC3623909
- DOI: 10.1371/journal.pone.0061009
Phylogenetic and structural diversity in the feline leukemia virus env gene
Abstract
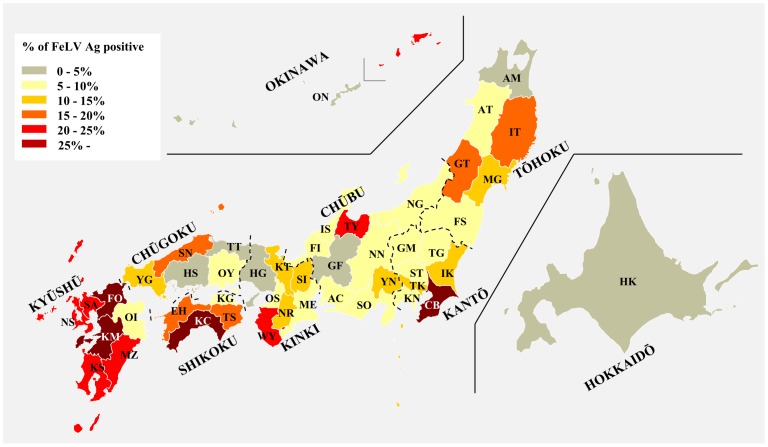
Feline leukemia virus (FeLV) belongs to the genus Gammaretrovirus, and causes a variety of neoplastic and non-neoplastic diseases in cats. Alteration of viral env sequences is thought to be associated with disease specificity, but the way in which genetic diversity of FeLV contributes to the generation of such variants in nature is poorly understood. We isolated FeLV env genes from naturally infected cats in Japan and analyzed the evolutionary dynamics of these genes. Phylogenetic reconstructions separated our FeLV samples into three distinct genetic clusters, termed Genotypes I, II, and III. Genotype I is a major genetic cluster and can be further classified into Clades 1-7 in Japan. Genotypes were correlated with geographical distribution; Genotypes I and II were distributed within Japan, whilst FeLV samples from outside Japan belonged to Genotype III. These results may be due to geographical isolation of FeLVs in Japan. The observed structural diversity of the FeLV env gene appears to be caused primarily by mutation, deletion, insertion and recombination, and these variants may be generated de novo in individual cats. FeLV interference assay revealed that FeLV genotypes did not correlate with known FeLV receptor subgroups. We have identified the genotypes which we consider to be reliable for evaluating phylogenetic relationships of FeLV, which embrace the high structural diversity observed in our sample. Overall, these findings extend our understanding of Gammaretrovirus evolutionary patterns in the field, and may provide a useful basis for assessing the emergence of novel strains and understanding the molecular mechanisms of FeLV transmission in cats.
Conflict of interest statement
Figures






Similar articles
-
Feline Leukemia Virus (FeLV) Endogenous and Exogenous Recombination Events Result in Multiple FeLV-B Subtypes during Natural Infection.J Virol. 2021 Aug 25;95(18):e0035321. doi: 10.1128/JVI.00353-21. Epub 2021 Aug 25. J Virol. 2021. PMID: 34232703 Free PMC article.
-
Genetic diversity in the feline leukemia virus gag gene.Virus Res. 2015 Jun 2;204:74-81. doi: 10.1016/j.virusres.2015.04.008. Epub 2015 Apr 17. Virus Res. 2015. PMID: 25892717
-
Reduced Folate Carrier: an Entry Receptor for a Novel Feline Leukemia Virus Variant.J Virol. 2019 Jun 14;93(13):e00269-19. doi: 10.1128/JVI.00269-19. Print 2019 Jul 1. J Virol. 2019. PMID: 30996094 Free PMC article.
-
Pathogenicity of feline leukemia virus is commonly associated with variant viruses.Leukemia. 1992;6 Suppl 3:153S-154S. Leukemia. 1992. PMID: 1318467 Review.
-
Endogenous env elements: partners in generation of pathogenic feline leukemia viruses.Virus Genes. 1995;11(2-3):147-61. doi: 10.1007/BF01728655. Virus Genes. 1995. PMID: 8828142 Review.
Cited by
-
Molecular characterization of bovine leukemia virus from Moldovan dairy cattle.Arch Virol. 2017 Jun;162(6):1563-1576. doi: 10.1007/s00705-017-3241-4. Epub 2017 Feb 17. Arch Virol. 2017. PMID: 28213870 Free PMC article.
-
Could Phylogenetic Analysis Be Used for Feline Leukemia Virus (FeLV) Classification?Viruses. 2022 Jan 26;14(2):249. doi: 10.3390/v14020249. Viruses. 2022. PMID: 35215842 Free PMC article.
-
Notch2 transduction by feline leukemia virus in a naturally infected cat.J Vet Med Sci. 2014 Apr;76(4):553-7. doi: 10.1292/jvms.13-0344. Epub 2013 Dec 9. J Vet Med Sci. 2014. PMID: 24317268 Free PMC article.
-
Feline Leukemia Virus (FeLV) Endogenous and Exogenous Recombination Events Result in Multiple FeLV-B Subtypes during Natural Infection.J Virol. 2021 Aug 25;95(18):e0035321. doi: 10.1128/JVI.00353-21. Epub 2021 Aug 25. J Virol. 2021. PMID: 34232703 Free PMC article.
-
Novel Feline Leukemia Virus Interference Group Based on the env Gene.J Virol. 2016 Apr 14;90(9):4832-4837. doi: 10.1128/JVI.03229-15. Print 2016 May. J Virol. 2016. PMID: 26889025 Free PMC article.
References
-
- Hisasue M, Nagashima N, Nishigaki K, Fukuzawa I, Ura S, et al. (2009) Myelodysplastic syndromes and acute myeloid leukemia in cats infected with feline leukemia virus clone33 containing a unique long terminal repeat. Int J Cancer 124: 1133–1141. - PubMed
-
- Neil JC, Fulton R, Rigby M, Stewart M (1991) Feline leukaemia virus: generation of pathogenic and oncogenic variants. Curr Top Microbiol Immunol 171: 67–93. - PubMed
-
- Matsumoto Y, Momoi Y, Watari T, Goitsuka R, Tsujimoto H, et al. (1992) Detection of enhancer repeats in the long terminal repeats of feline leukemia viruses from cats with spontaneous neoplastic and nonneoplastic diseases. Virology 189: 745–749. - PubMed
-
- Miura T, Shibuya M, Tsujimoto H, Fukasawa M, Hayami M (1989) Molecular cloning of a feline leukemia provirus integrated adjacent to the c-myc gene in a feline T-cell leukemia cell line and the unique structure of its long terminal repeat. Virology 169: 458–461. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
