

Sulfatide regulates caspase-3-independent apoptosis of influenza A virus through viral PB1-F2 protein
- PMID: 23593400
- PMCID: PMC3617187
- DOI: 10.1371/journal.pone.0061092
Sulfatide regulates caspase-3-independent apoptosis of influenza A virus through viral PB1-F2 protein
Abstract
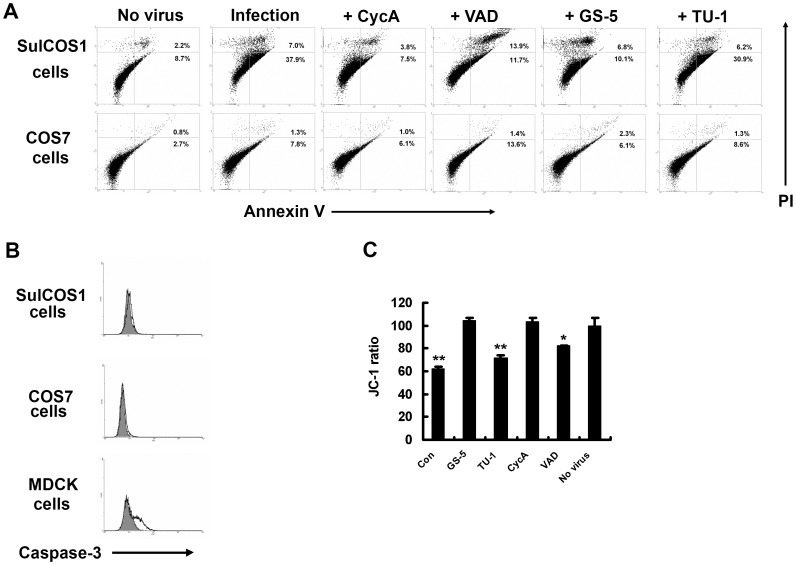
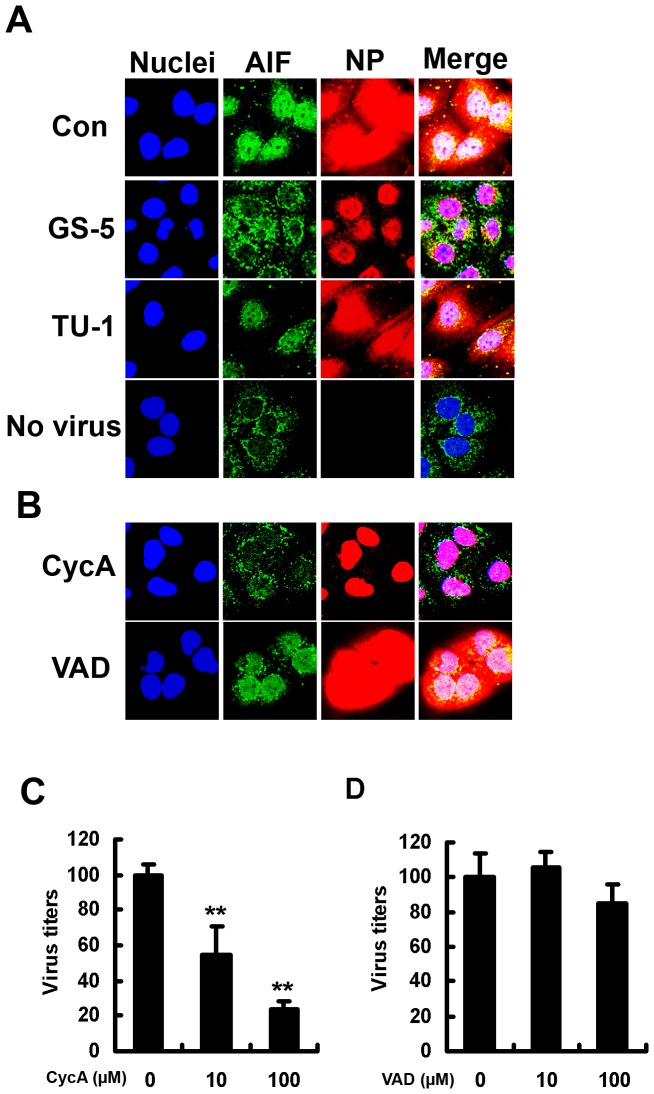
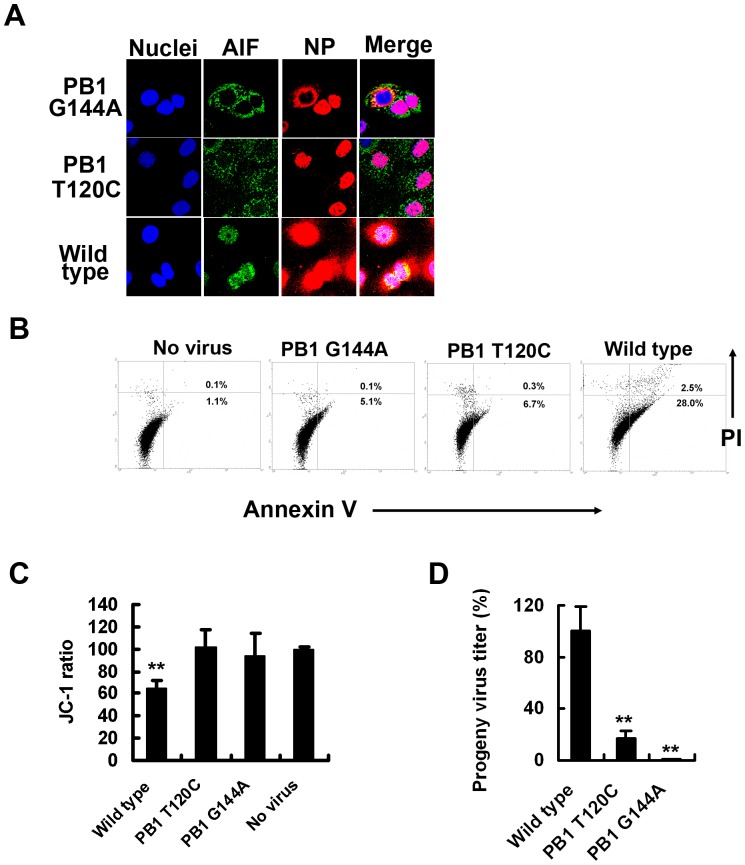
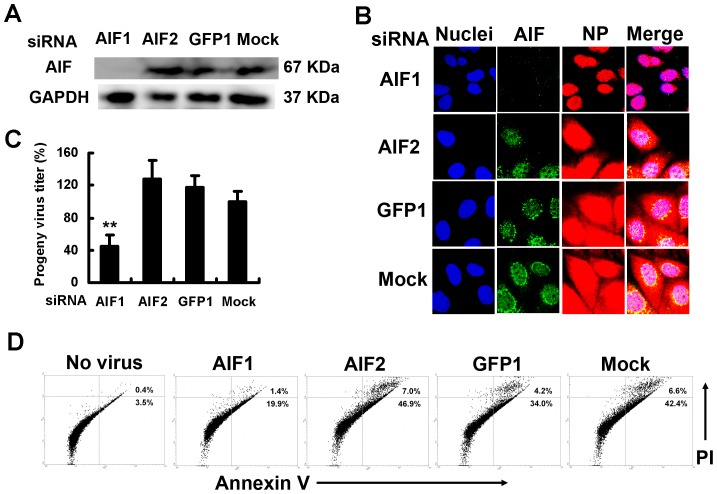
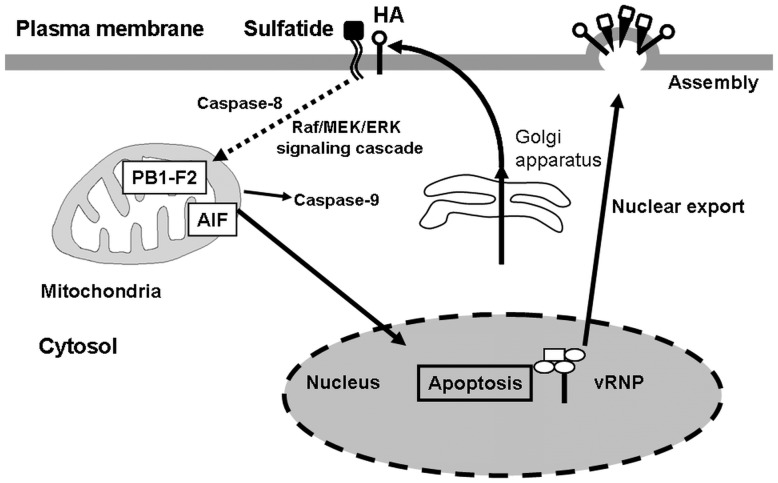
Influenza A virus (IAV) generally causes caspase-dependent apoptosis based on caspase-3 activation, resulting in nuclear export of newly synthesized viral nucleoprotein (NP) and elevated virus replication. Sulfatide, a sulfated galactosylsphingolipid, enhances IAV replication through promoting newly synthesized viral NP export induced by association of sulfatide with hemagglutinin delivered to the cell surface. Here, we demonstrated that sulfatide is involved in caspase-3-independent apoptosis initiated by the PB1-F2 protein of IAV by using genetically sulfatide-produced cells and PB1-F2-deficient IAVs. Sulfatide-deficient COS7 cells showed no virus-induced apoptosis, whereas SulCOS1 cells, sulfatide-enriched COS7 cells that genetically expressed the two transferases required for sulfatide synthesis from ceramide, showed an increase in IAV replication and were susceptible to caspase-3-independent apoptosis. Additionally, PB1-F2-deficient IAVs, which were generated by using a plasmid-based reverse genetics system from a genetic background of A/WSN/33 (H1N1), demonstrated that PB1-F2 contributed to caspase-3-independent apoptosis in IAV-infected SulCOS1 cells. Our results show that sulfatide plays a critical role in efficient IAV propagation via caspase-3-independent apoptosis initiated by the PB1-F2 protein.
Conflict of interest statement
Figures
Similar articles
-
Sulfatide is required for efficient replication of influenza A virus.J Virol. 2008 Jun;82(12):5940-50. doi: 10.1128/JVI.02496-07. Epub 2008 Apr 16. J Virol. 2008. PMID: 18417587 Free PMC article.
-
Role of sulfatide in influenza A virus replication.Biol Pharm Bull. 2015;38(6):809-16. doi: 10.1248/bpb.b15-00119. Biol Pharm Bull. 2015. PMID: 26027821 Review.
-
NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein.Proc Natl Acad Sci U S A. 2014 May 20;111(20):E2110-9. doi: 10.1073/pnas.1322118111. Epub 2014 May 5. Proc Natl Acad Sci U S A. 2014. PMID: 24799673 Free PMC article.
-
H5N1 Influenza A Virus PB1-F2 Relieves HAX-1-Mediated Restriction of Avian Virus Polymerase PA in Human Lung Cells.J Virol. 2018 May 14;92(11):e00425-18. doi: 10.1128/JVI.00425-18. Print 2018 Jun 1. J Virol. 2018. PMID: 29563290 Free PMC article.
-
Influenza a virus PB1-F2 protein.Acta Virol. 2007;51(2):101-8. Acta Virol. 2007. PMID: 17900216 Review.
Cited by
-
Functional Analysis of Sulfatide in Influenza A Virus Infection and Replication.Methods Mol Biol. 2022;2556:97-122. doi: 10.1007/978-1-0716-2635-1_9. Methods Mol Biol. 2022. PMID: 36175630
-
Imaging of influenza virus sialidase activity in living cells.Sci Rep. 2014 May 2;4:4877. doi: 10.1038/srep04877. Sci Rep. 2014. PMID: 24786761 Free PMC article.
-
The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection.Nat Commun. 2020 Jan 9;11(1):162. doi: 10.1038/s41467-019-13974-w. Nat Commun. 2020. PMID: 31919357 Free PMC article.
-
Identification of virulence determinants in influenza viruses.Anal Chem. 2014 Jul 15;86(14):6911-7. doi: 10.1021/ac500659f. Epub 2014 Jun 27. Anal Chem. 2014. PMID: 24937567 Free PMC article.
-
Preliminary study of Yinhuapinggan granule against H1N1 influenza virus infection in mice through inhibition of apoptosis.Pharm Biol. 2020 Dec;58(1):979-991. doi: 10.1080/13880209.2020.1818792. Pharm Biol. 2020. PMID: 32962483 Free PMC article.
References
-
- Takizawa T, Tatematsu C, Ohashi K, Nakanishi Y (1999) Recruitment of apoptotic cysteine proteases (caspases) in influenza virus-induced cell death. Microbiol Immunol 43: 245–252. - PubMed
-
- Morris SJ, Price GE, Barnett JM, Hiscox SA, Smith H, et al. (1999) Role of neuraminidase in influenza virus-induced apoptosis. J Gen Virol 80: 137–146. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
