

Understanding the folding-function tradeoff in proteins
- PMID: 23593437
- PMCID: PMC3625163
- DOI: 10.1371/journal.pone.0061222
Understanding the folding-function tradeoff in proteins
Abstract
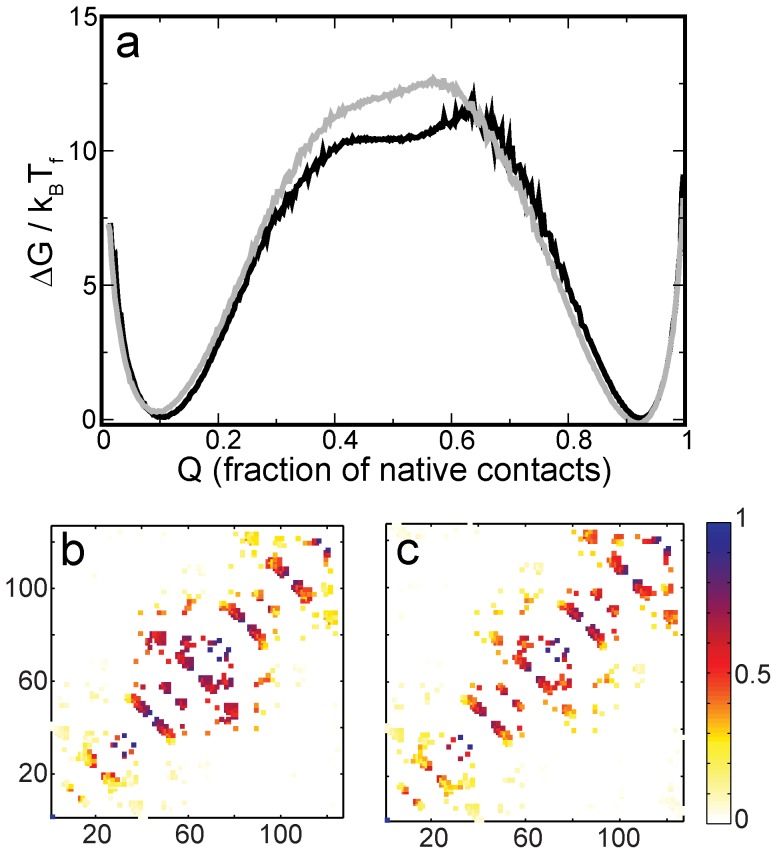
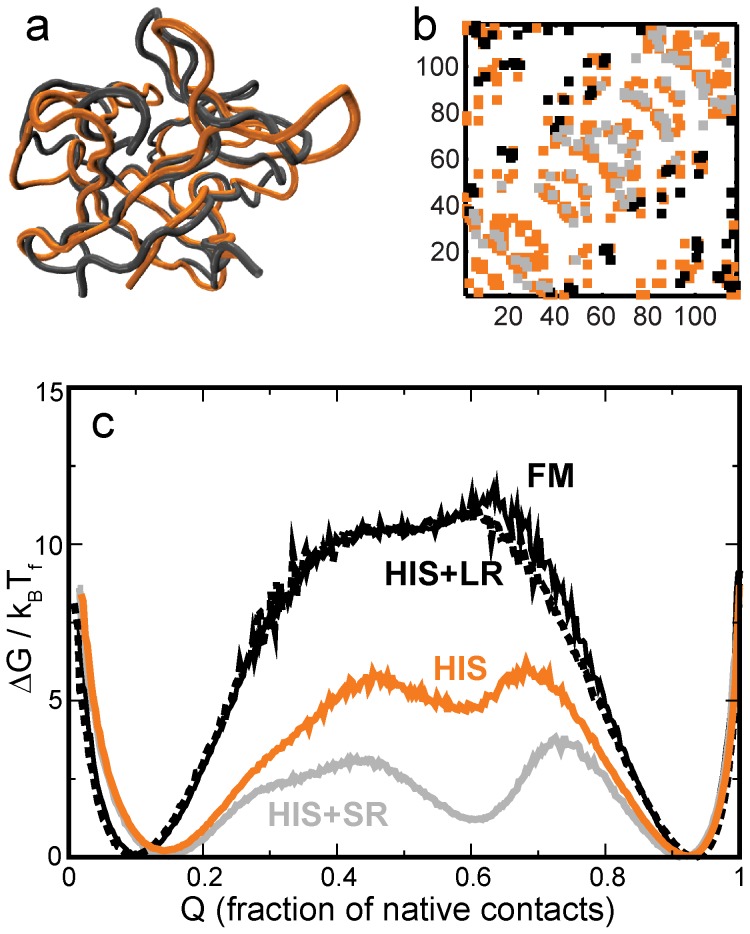
When an amino-acid sequence cannot be optimized for both folding and function, folding can get compromised in favor of function. To understand this tradeoff better, we devise a novel method for extracting the "function-less" folding-motif of a protein fold from a set of structurally similar but functionally diverse proteins. We then obtain the β-trefoil folding-motif, and study its folding using structure-based models and molecular dynamics simulations. CompariA protein sequence serves two purpson with the folding of wild-type β-trefoil proteins shows that function affects folding in two ways: In the slower folding interleukin-1β, binding sites make the fold more complex, increase contact order and slow folding. In the faster folding hisactophilin, residues which could have been part of the folding-motif are used for function. This reduces the density of native contacts in functional regions and increases folding rate. The folding-motif helps identify subtle structural deviations which perturb folding. These may then be used for functional annotation. Further, the folding-motif could potentially be used as a first step in the sequence design of function-less scaffold proteins. Desired function can then be engineered into these scaffolds.
Conflict of interest statement
Figures






References
-
- Kloczkowski A, Sen TZ, Jernigan RL (2005) Promiscuous vs. native protein function. Insights from studying collective motions in proteins with elastic network models. J Biomol Struct Dyn 22: 621–624 and references therein.
-
- Frauenfelder H, Sligar SG, Wolynes PG (1991) The energy landscapes and motions of proteins. Science 254: 1598–1603. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
