

New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications
- PMID: 23593929
- PMCID: PMC3663883
- DOI: 10.1111/petr.12076
New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications
Abstract
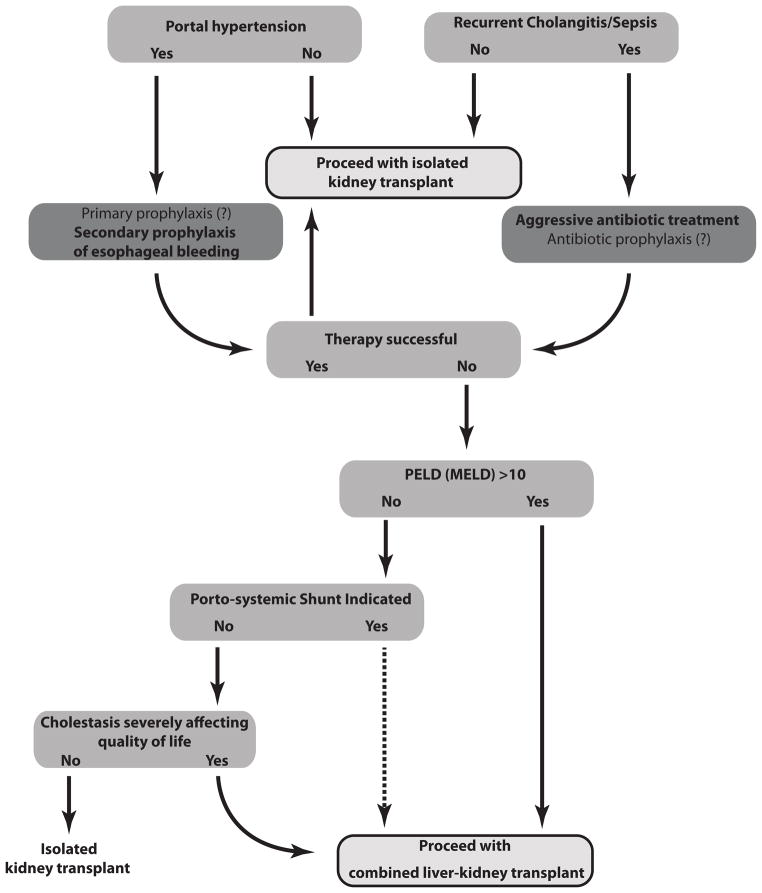
Improved neonatal medical care and renal replacement technology have improved the long-term survival of patients with ARPKD. Ten-yr survival of those surviving the first year of life is reported to be 82% and is continuing to improve further. However, despite increases in overall survival and improved treatment of systemic hypertension and other complications of their renal disease, nearly 50% of survivors will develop ESRD within the first decade of life. In addition to renal pathology, patients with ARPKD develop ductal plate malformations with cystic dilation of intra- and extrahepatic bile ducts resulting in CHF and Caroli syndrome. Many patients with CHF will develop portal hypertension with resulting esophageal varices, splenomegaly, hypersplenism, protein losing enteropathy, and gastrointestinal bleeding. Management of portal hypertension may require EBL of esophageal varices or porto-systemic shunting. Complications of hepatic involvement can include ascending cholangitis, cholestasis with malabsorption of fat-soluble vitamins, and rarely benign or malignant liver tumors. Patients with ARPKD who eventually reach ESRD, and ultimately require kidney transplantation, present a unique set of complications related to their underlying hepato-biliary disease. In this review, we focus on new approaches to these challenging patients, including the indications for liver transplantation in ARPKD patients with severe chronic kidney disease awaiting kidney transplant. While survival in patients with ARPKD and isolated kidney transplant is comparable to that of age-matched pediatric patients who have received kidney transplants due to other primary renal diseases, 64-80% of the mortality occurring in ARPKD kidney transplant patients is attributed to cholangitis/sepsis, which is related to their hepato-biliary disease. Recent data demonstrate that surgical mortality among pediatric liver transplant recipients is decreased to <10% at one yr. The immunosuppressive regimen used for kidney transplant recipients is adequate for most liver transplant recipients. We therefore suggest that in a select group of ARPKD patients with recurrent cholangitis or complications of portal hypertension, combined liver-kidney transplant is a viable option. Although further study is necessary to confirm our approach, we believe that combined liver-kidney transplantation can potentially decrease overall mortality and morbidity in carefully selected ARPKD patients with ESRD and clinically significant CHF.
© 2013 John Wiley & Sons A/S.
Similar articles
-
Phenotypic variation and long-term outcome in children with congenital hepatic fibrosis.J Pediatr Gastroenterol Nutr. 2013 Aug;57(2):161-6. doi: 10.1097/MPG.0b013e318291e72b. J Pediatr Gastroenterol Nutr. 2013. PMID: 23518487
-
Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease.J Pediatr Gastroenterol Nutr. 2012 May;54(5):580-7. doi: 10.1097/MPG.0b013e31824711b7. J Pediatr Gastroenterol Nutr. 2012. PMID: 22197937 Free PMC article. Review.
-
Morbidity from congenital hepatic fibrosis after renal transplantation for autosomal recessive polycystic kidney disease.Am J Transplant. 2002 Apr;2(4):360-5. doi: 10.1034/j.1600-6143.2002.20412.x. Am J Transplant. 2002. PMID: 12118859
-
Long-term kidney and liver outcome in 50 children with autosomal recessive polycystic kidney disease.Pediatr Nephrol. 2021 May;36(5):1165-1173. doi: 10.1007/s00467-020-04808-9. Epub 2020 Nov 9. Pediatr Nephrol. 2021. PMID: 33165639
-
Liver disease in autosomal recessive polycystic kidney disease.Pediatr Transplant. 2005 Oct;9(5):634-9. doi: 10.1111/j.1399-3046.2005.00342.x. Pediatr Transplant. 2005. PMID: 16176423 Review.
Cited by
-
Autosomal Recessive Polycystic Kidney Disease-The Clinical Aspects and Diagnostic Challenges.J Pediatr Genet. 2021 Mar;10(1):1-8. doi: 10.1055/s-0040-1714701. Epub 2020 Jul 29. J Pediatr Genet. 2021. PMID: 33552631 Free PMC article. Review.
-
Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects.Pediatrics. 2014 Sep;134(3):e833-45. doi: 10.1542/peds.2013-3646. Epub 2014 Aug 11. Pediatrics. 2014. PMID: 25113295 Free PMC article. Review.
-
Transplantation in autosomal recessive polycystic kidney disease: liver and/or kidney?Pediatr Nephrol. 2015 Aug;30(8):1233-42. doi: 10.1007/s00467-014-2887-3. Epub 2014 Aug 13. Pediatr Nephrol. 2015. PMID: 25115876 Review.
-
Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy.Pediatr Res. 2014 Jan;75(1-2):148-57. doi: 10.1038/pr.2013.191. Epub 2013 Oct 31. Pediatr Res. 2014. PMID: 24336431 Free PMC article. Review.
-
A teenage patient with autosomal recessive polycystic kidney disease, a splenorenal shunt, and congenital hepatic fibrosis: a case report.J Bras Nefrol. 2019 Apr-Jun;41(2):300-303. doi: 10.1590/2175-8239-jbn-2018-0081. Epub 2018 Sep 6. J Bras Nefrol. 2019. PMID: 30199558 Free PMC article.
References
-
- ROY S, DILLIN MJ, TROMPETER RS, BARRATT TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302–306. - PubMed
-
- ADEVA M, EL-YOUSSEF M, ROSETTI S, KAMATH PS, KUBLY V, CONSUGAR MB, MILLINER DM, KING BF, TORRES VE, HARRIS PC. Clinical and molecular characterisations defines a broadened spectrum of autosomal recessive polycystic disease (ARPKD) Medicine. 2006;85:1–21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
