

Population genetics of rare variants and complex diseases
- PMID: 23594490
- PMCID: PMC3698246
- DOI: 10.1159/000346826
Population genetics of rare variants and complex diseases
Abstract
Objectives: Identifying drivers of complex traits from the noisy signals of genetic variation obtained from high-throughput genome sequencing technologies is a central challenge faced by human geneticists today. We hypothesize that the variants involved in complex diseases are likely to exhibit non-neutral evolutionary signatures. Uncovering the evolutionary history of all variants is therefore of intrinsic interest for complex disease research. However, doing so necessitates the simultaneous elucidation of the targets of natural selection and population-specific demographic history.
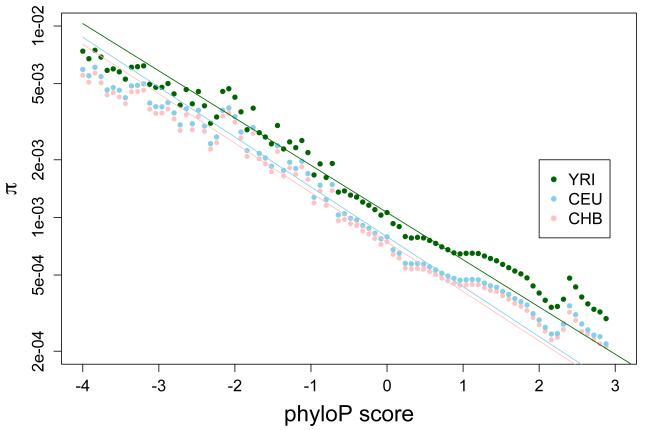
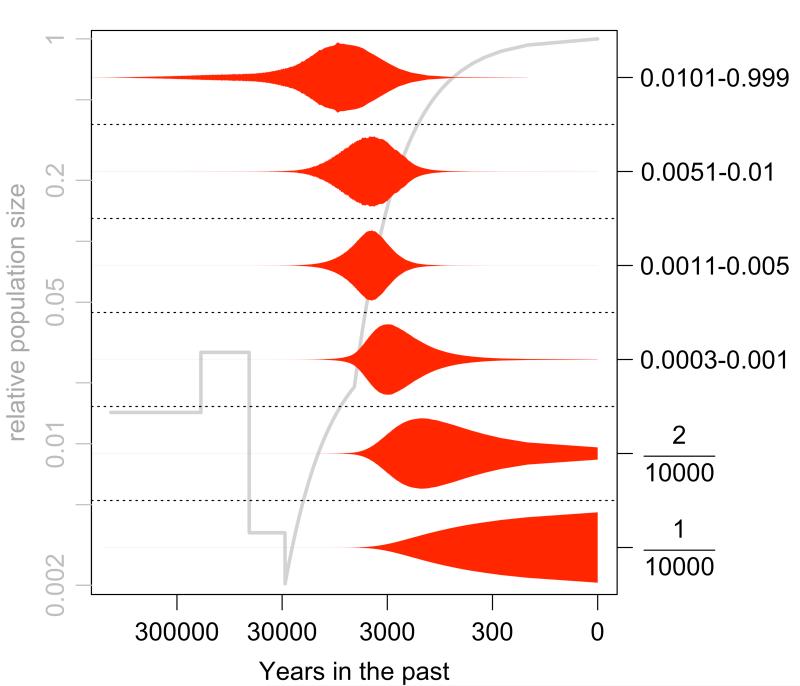
Methods: Here we characterize the action of natural selection operating across complex disease categories, and use population genetic simulations to evaluate the expected patterns of genetic variation in large samples. We focus on populations that have experienced historical bottlenecks followed by explosive growth (consistent with many human populations), and describe the differences between evolutionarily deleterious mutations and those that are neutral.
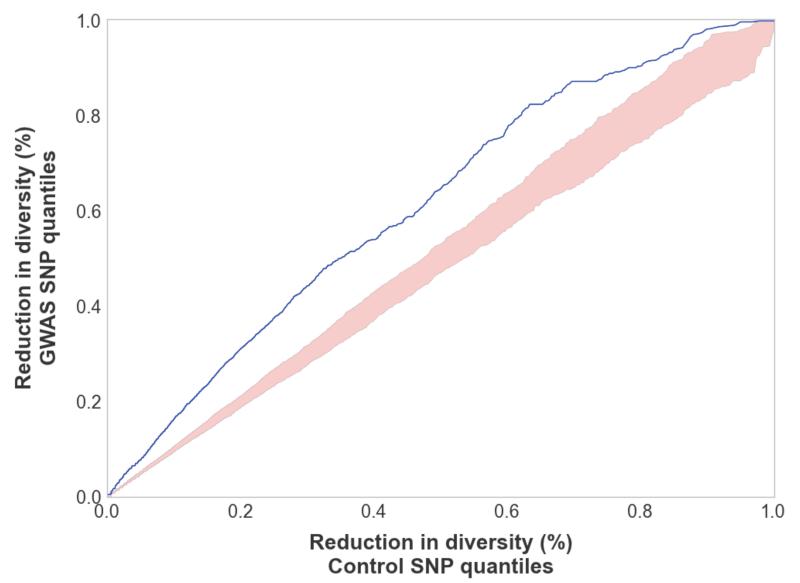
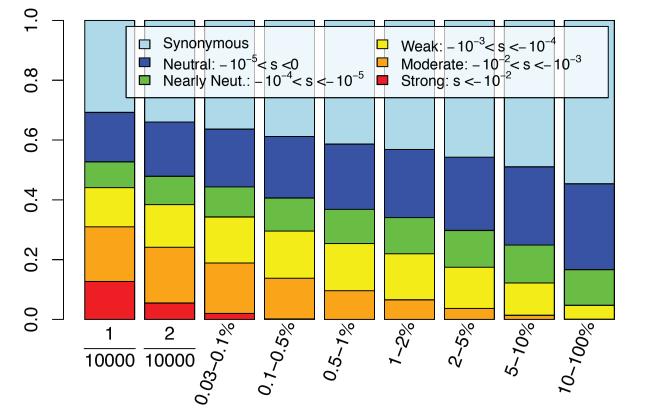
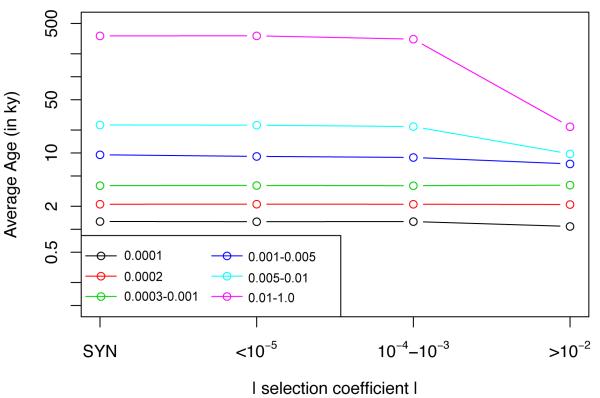
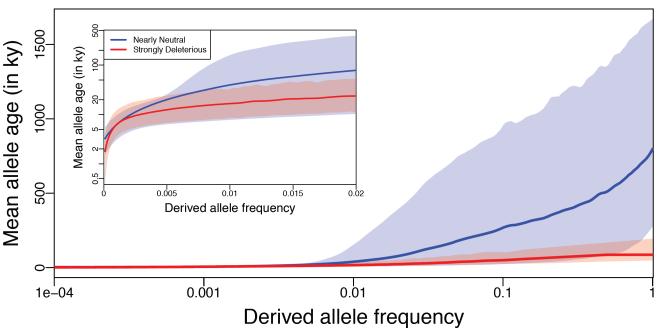
Results: Genes associated with several complex disease categories exhibit stronger signatures of purifying selection than non-disease genes. In addition, loci identified through genome-wide association studies of complex traits also exhibit signatures consistent with being in regions recurrently targeted by purifying selection. Through simulations, we show that population bottlenecks and rapid growth enable deleterious rare variants to persist at low frequencies just as long as neutral variants, but low-frequency and common variants tend to be much younger than neutral variants. This has resulted in a large proportion of modern-day rare alleles that have a deleterious effect on function and that potentially contribute to disease susceptibility.
Conclusions: The key question for sequencing-based association studies of complex traits is how to distinguish between deleterious and benign genetic variation. We used population genetic simulations to uncover patterns of genetic variation that distinguish these two categories, especially derived allele age, thereby providing inroads into novel methods for characterizing rare genetic variation driving complex diseases.
Copyright © 2013 S. Karger AG, Basel.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
