

Acute suppression of spontaneous neurotransmission drives synaptic potentiation
- PMID: 23595756
- PMCID: PMC3661220
- DOI: 10.1523/JNEUROSCI.4998-12.2013
Acute suppression of spontaneous neurotransmission drives synaptic potentiation
Abstract
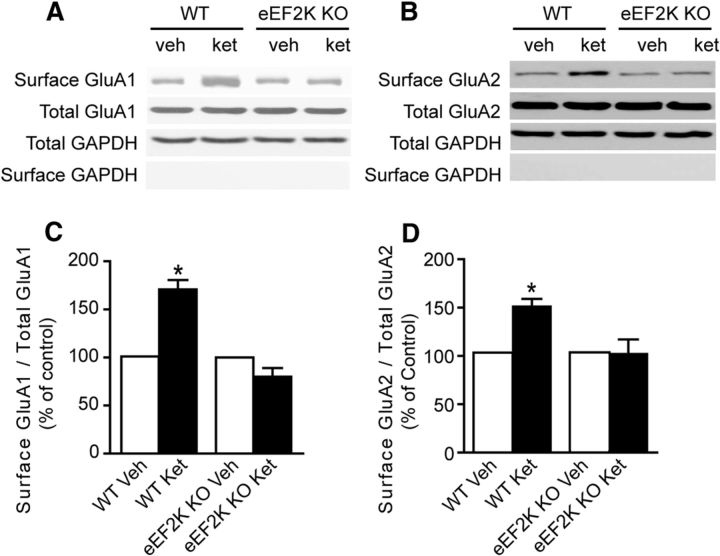
The impact of spontaneous neurotransmission on neuronal plasticity remains poorly understood. Here, we show that acute suppression of spontaneous NMDA receptor-mediated (NMDAR-mediated) neurotransmission potentiates synaptic responses in the CA1 regions of rat and mouse hippocampus. This potentiation requires protein synthesis, brain-derived neurotrophic factor expression, eukaryotic elongation factor-2 kinase function, and increased surface expression of AMPA receptors. Our behavioral studies link this same synaptic signaling pathway to the fast-acting antidepressant responses elicited by ketamine. We also show that selective neurotransmitter depletion from spontaneously recycling vesicles triggers synaptic potentiation via the same pathway as NMDAR blockade, demonstrating that presynaptic impairment of spontaneous release, without manipulation of evoked neurotransmission, is sufficient to elicit postsynaptic plasticity. These findings uncover an unexpectedly dynamic impact of spontaneous glutamate release on synaptic efficacy and provide new insight into a key synaptic substrate for rapid antidepressant action.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous