

Targeted exome sequencing of suspected mitochondrial disorders
- PMID: 23596069
- PMCID: PMC3719425
- DOI: 10.1212/WNL.0b013e3182918c40
Targeted exome sequencing of suspected mitochondrial disorders
Abstract
Objective: To evaluate the utility of targeted exome sequencing for the molecular diagnosis of mitochondrial disorders, which exhibit marked phenotypic and genetic heterogeneity.
Methods: We considered a diverse set of 102 patients with suspected mitochondrial disorders based on clinical, biochemical, and/or molecular findings, and whose disease ranged from mild to severe, with varying age at onset. We sequenced the mitochondrial genome (mtDNA) and the exons of 1,598 nuclear-encoded genes implicated in mitochondrial biology, mitochondrial disease, or monogenic disorders with phenotypic overlap. We prioritized variants likely to underlie disease and established molecular diagnoses in accordance with current clinical genetic guidelines.
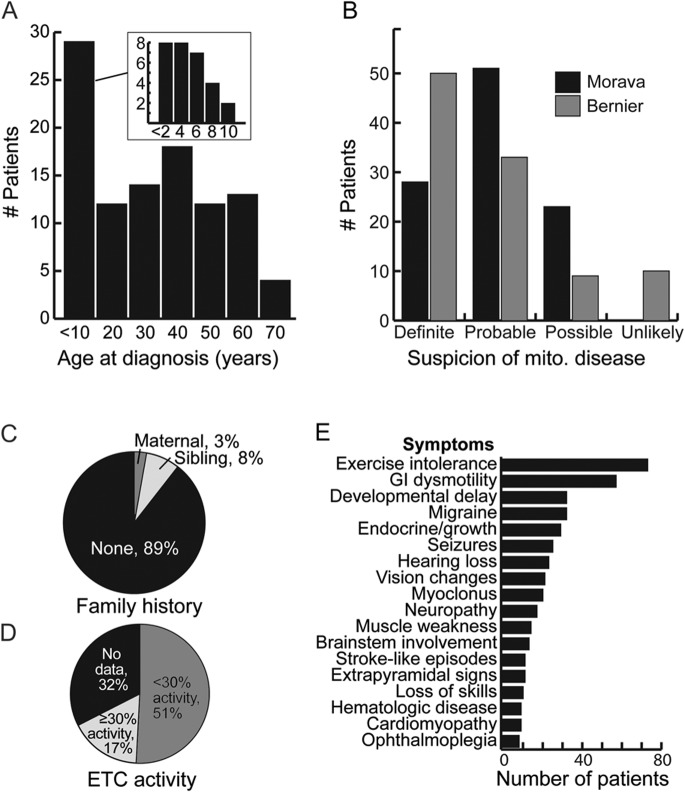
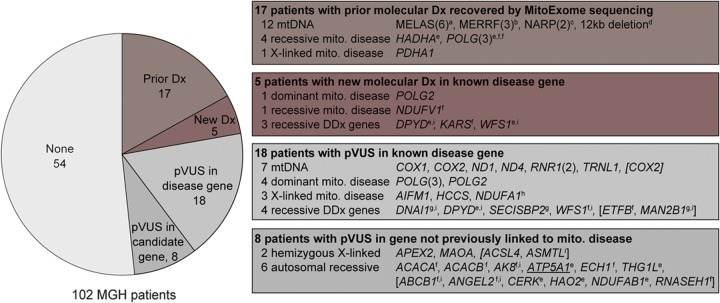
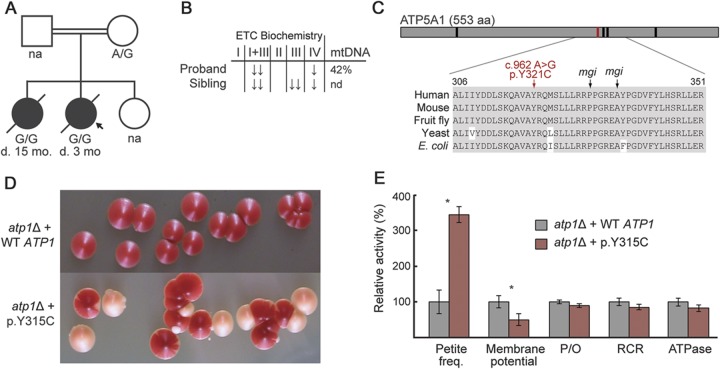
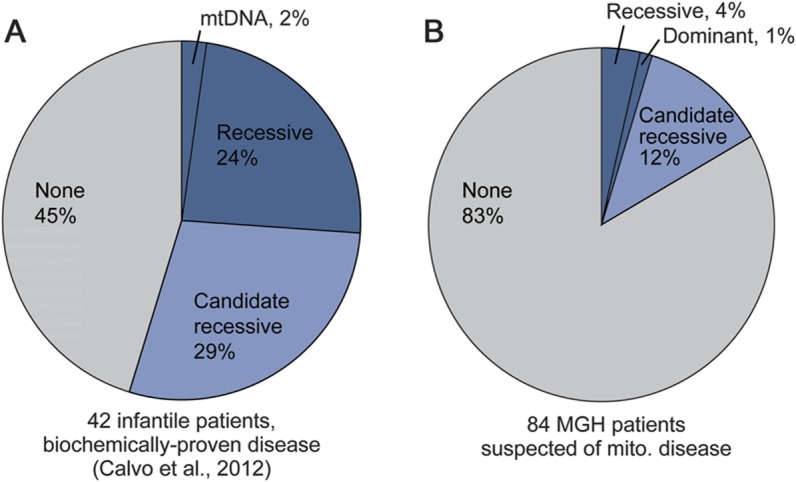
Results: Targeted exome sequencing yielded molecular diagnoses in established disease loci in 22% of cases, including 17 of 18 (94%) with prior molecular diagnoses and 5 of 84 (6%) without. The 5 new diagnoses implicated 2 genes associated with canonical mitochondrial disorders (NDUFV1, POLG2), and 3 genes known to underlie other neurologic disorders (DPYD, KARS, WFS1), underscoring the phenotypic and biochemical overlap with other inborn errors. We prioritized variants in an additional 26 patients, including recessive, X-linked, and mtDNA variants that were enriched 2-fold over background and await further support of pathogenicity. In one case, we modeled patient mutations in yeast to provide evidence that recessive mutations in ATP5A1 can underlie combined respiratory chain deficiency.
Conclusion: The results demonstrate that targeted exome sequencing is an effective alternative to the sequential testing of mtDNA and individual nuclear genes as part of the investigation of mitochondrial disease. Our study underscores the ongoing challenge of variant interpretation in the clinical setting.
Figures
References
-
- DiMauro S, Hirano M, Schon EA. Mitochondrial Medicine. New York: Informa Healthcare; 2006
-
- Chinnery PF. Mitochondrial disorders overview. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle: University of Washington; 1993
-
- Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 2012;366:1132–1141 - PubMed
-
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature 2012;491:374–383 - PubMed
-
- Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med 2005;37:222–232 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous