

Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation
- PMID: 23599344
- PMCID: PMC3650227
- DOI: 10.1101/gad.203984.112
Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation
Abstract
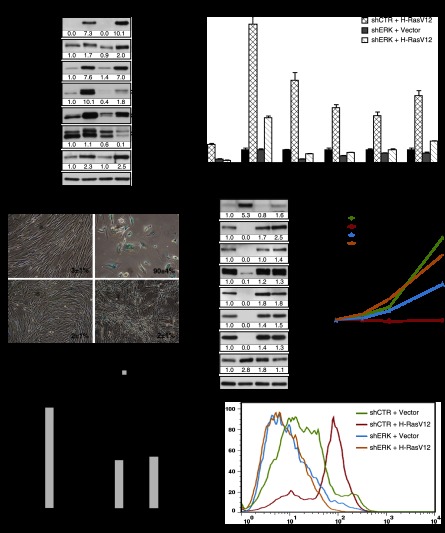
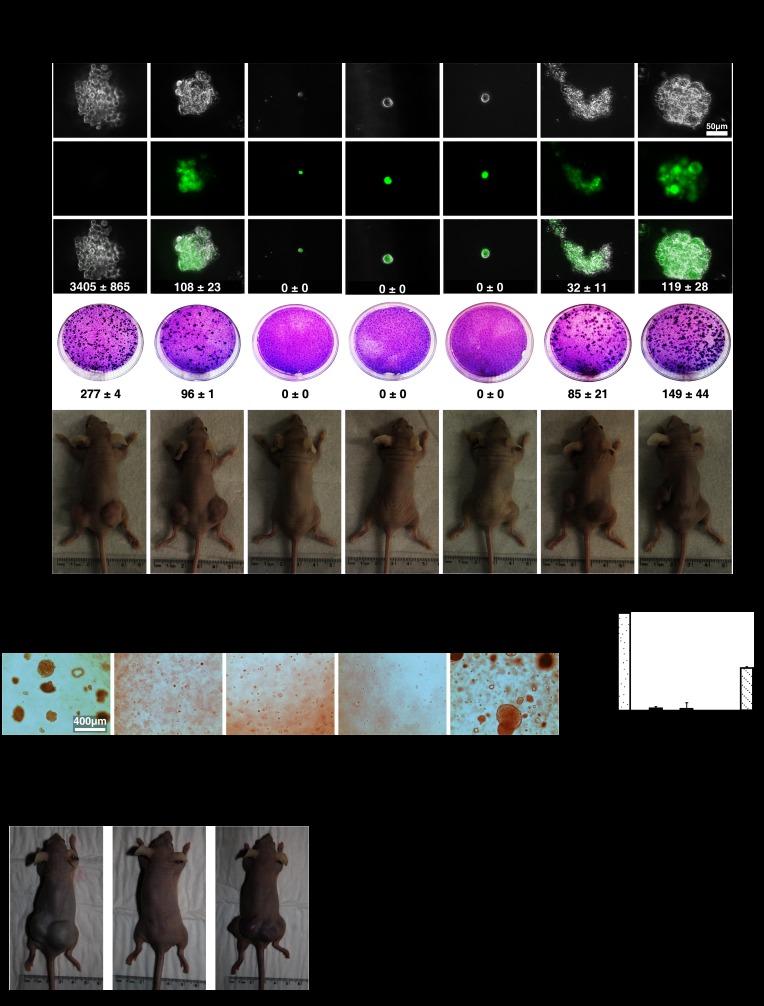
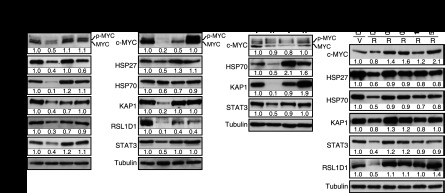
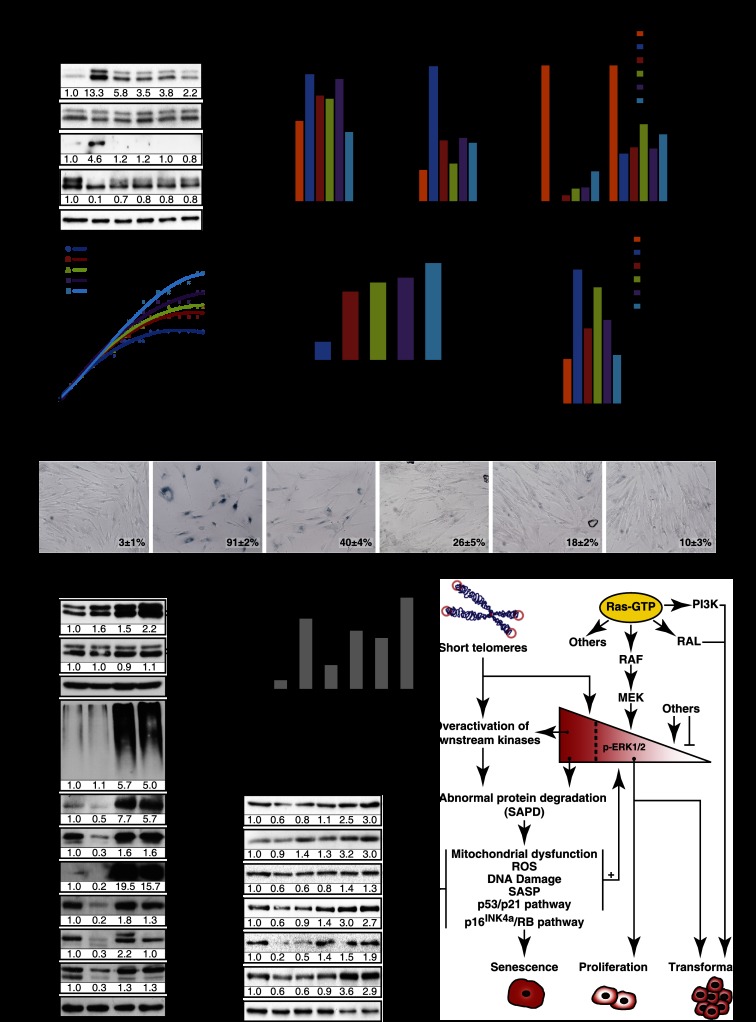
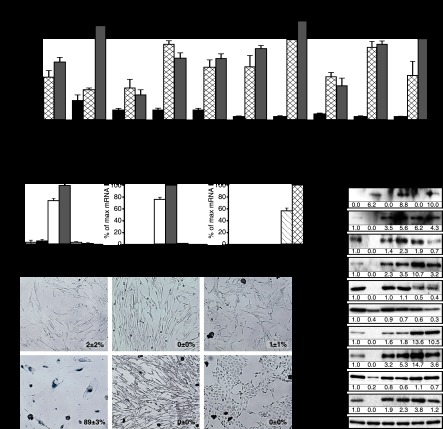
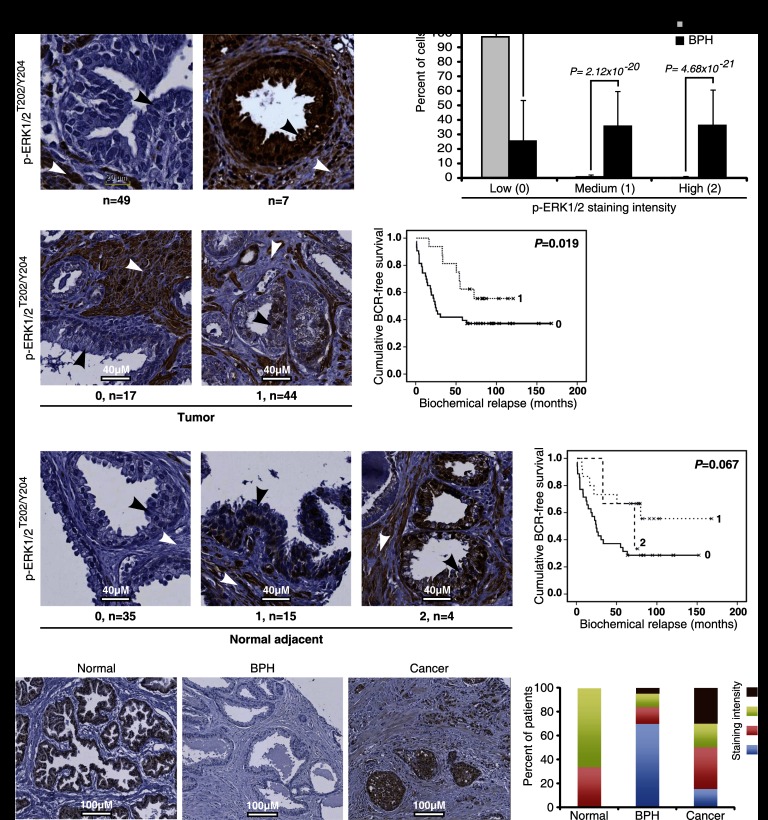
Constitutive activation of growth factor signaling pathways paradoxically triggers a cell cycle arrest known as cellular senescence. In primary cells expressing oncogenic ras, this mechanism effectively prevents cell transformation. Surprisingly, attenuation of ERK/MAP kinase signaling by genetic inactivation of Erk2, RNAi-mediated knockdown of ERK1 or ERK2, or MEK inhibitors prevented the activation of the senescence mechanism, allowing oncogenic ras to transform primary cells. Mechanistically, ERK-mediated senescence involved the proteasome-dependent degradation of proteins required for cell cycle progression, mitochondrial functions, cell migration, RNA metabolism, and cell signaling. This senescence-associated protein degradation (SAPD) was observed not only in cells expressing ectopic ras, but also in cells that senesced due to short telomeres. Individual RNAi-mediated inactivation of SAPD targets was sufficient to restore senescence in cells transformed by oncogenic ras or trigger senescence in normal cells. Conversely, the anti-senescence viral oncoproteins E1A, E6, and E7 prevented SAPD. In human prostate neoplasms, high levels of phosphorylated ERK were found in benign lesions, correlating with other senescence markers and low levels of STAT3, one of the SAPD targets. We thus identified a mechanism that links aberrant activation of growth signaling pathways and short telomeres to protein degradation and cellular senescence.
Figures


References
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 - PubMed
-
- Bostwick DG 1996. Prospective origins of prostate carcinoma. Prostatic intraepithelial neoplasia and atypical adenomatous hyperplasia. Cancer 78: 330–336 - PubMed
-
- Bostwick DG, Cooner WH, Denis L, Jones GW, Scardino PT, Murphy GP 1992. The association of benign prostatic hyperplasia and cancer of the prostate. Cancer 70: 291–301 - PubMed
-
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous