

LocARNAscan: Incorporating thermodynamic stability in sequence and structure-based RNA homology search
- PMID: 23601347
- PMCID: PMC3716875
- DOI: 10.1186/1748-7188-8-14
LocARNAscan: Incorporating thermodynamic stability in sequence and structure-based RNA homology search
Abstract
Background: The search for distant homologs has become an import issue in genome annotation. A particular difficulty is posed by divergent homologs that have lost recognizable sequence similarity. This same problem also arises in the recognition of novel members of large classes of RNAs such as snoRNAs or microRNAs that consist of families unrelated by common descent. Current homology search tools for structured RNAs are either based entirely on sequence similarity (such as blast or hmmer) or combine sequence and secondary structure. The most prominent example of the latter class of tools is Infernal. Alternatives are descriptor-based methods. In most practical applications published to-date, however, the information contained in covariance models or manually prescribed search patterns is dominated by sequence information. Here we ask two related questions: (1) Is secondary structure alone informative for homology search and the detection of novel members of RNA classes? (2) To what extent is the thermodynamic propensity of the target sequence to fold into the correct secondary structure helpful for this task?
Results: Sequence-structure alignment can be used as an alternative search strategy. In this scenario, the query consists of a base pairing probability matrix, which can be derived either from a single sequence or from a multiple alignment representing a set of known representatives. Sequence information can be optionally added to the query. The target sequence is pre-processed to obtain local base pairing probabilities. As a search engine we devised a semi-global scanning variant of LocARNA's algorithm for sequence-structure alignment. The LocARNAscan tool is optimized for speed and low memory consumption. In benchmarking experiments on artificial data we observe that the inclusion of thermodynamic stability is helpful, albeit only in a regime of extremely low sequence information in the query. We observe, furthermore, that the sensitivity is bounded in particular by the limited accuracy of the predicted local structures of the target sequence.
Conclusions: Although we demonstrate that a purely structure-based homology search is feasible in principle, it is unlikely to outperform tools such as Infernal in most application scenarios, where a substantial amount of sequence information is typically available. The LocARNAscan approach will profit, however, from high throughput methods to determine RNA secondary structure. In transcriptome-wide applications, such methods will provide accurate structure annotations on the target side.
Availability: Source code of the free software LocARNAscan 1.0 and supplementary data are available at http://www.bioinf.uni-leipzig.de/Software/LocARNAscan.
Figures
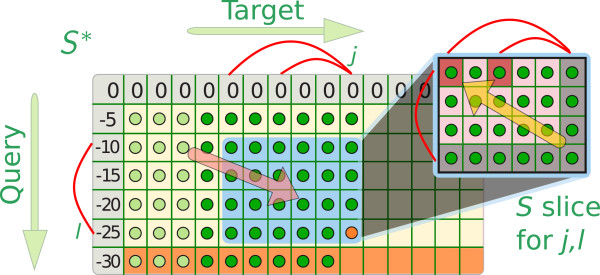
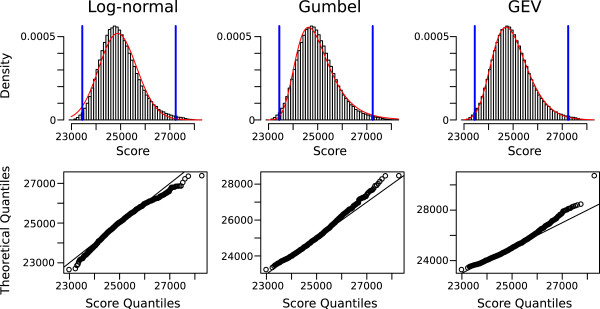
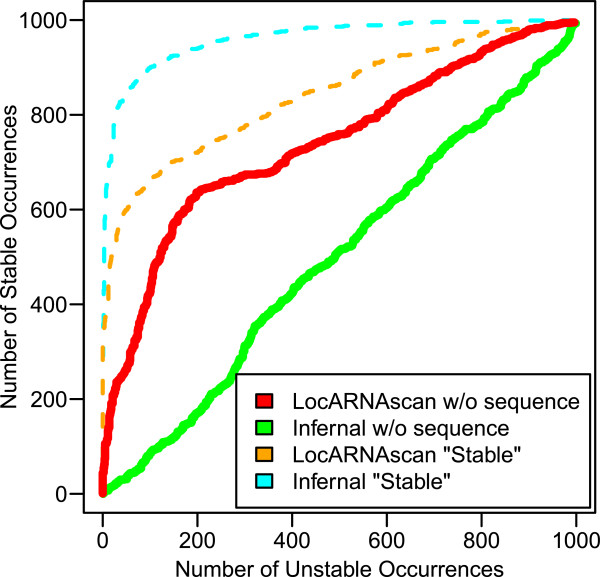
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
