

Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation
- PMID: 23603814
- PMCID: PMC3899792
- DOI: 10.1038/nm.3111
Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation
Abstract
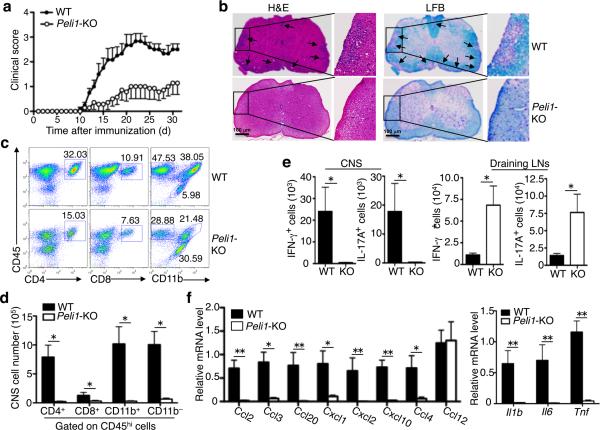
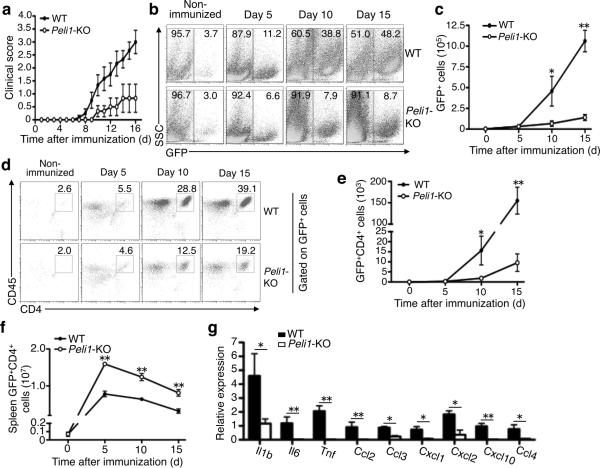
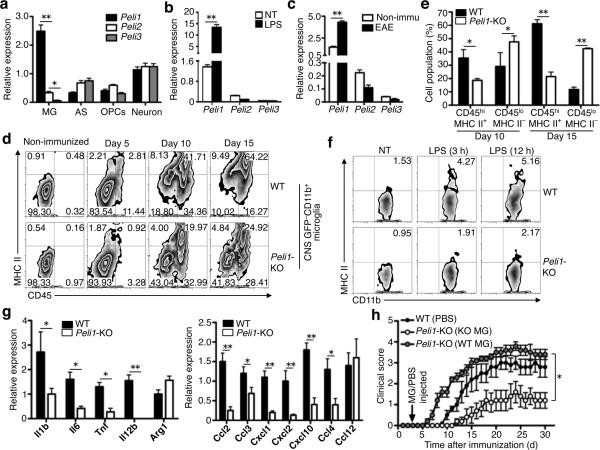
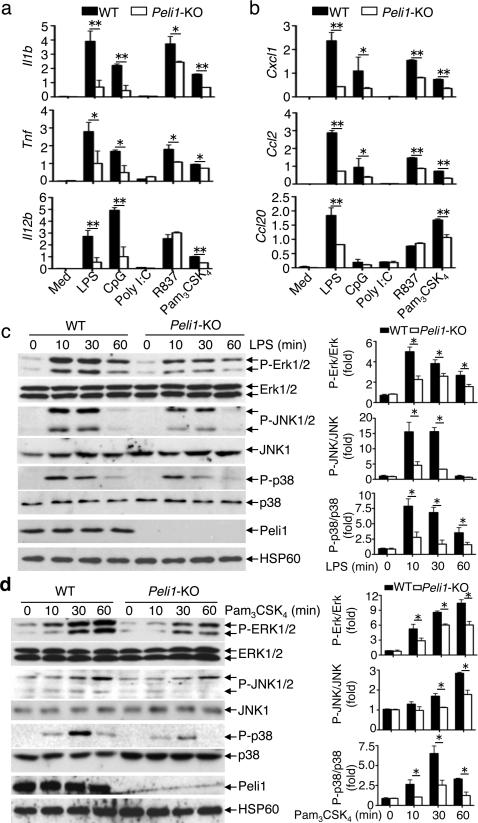
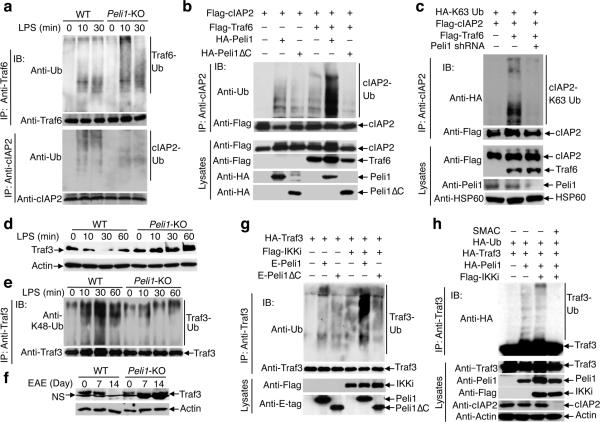
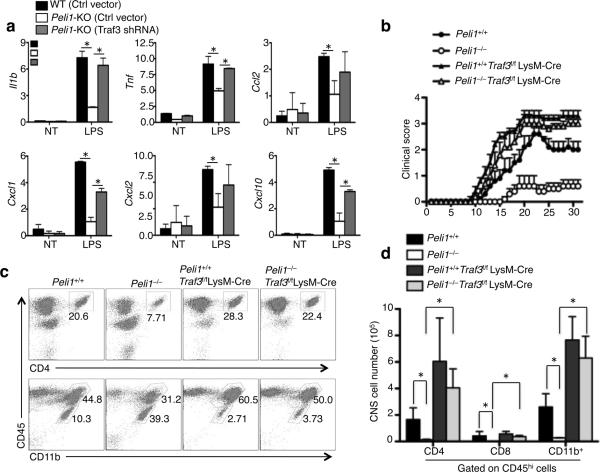
Microglia are crucial for the pathogenesis of multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis (EAE). Here we show that the E3 ubiquitin ligase Peli1 is abundantly expressed in microglia and promotes microglial activation during the course of EAE induction. Peli1 mediates the induction of chemokines and proinflammatory cytokines in microglia and thereby promotes recruitment of T cells into the central nervous system. The severity of EAE is reduced in Peli1-deficient mice despite their competent induction of inflammatory T cells in the peripheral lymphoid organs. Notably, Peli1 regulates Toll-like receptor (TLR) pathway signaling by promoting degradation of TNF receptor-associated factor 3 (Traf3), a potent inhibitor of mitogen-activated protein kinase (MAPK) activation and gene induction. Ablation of Traf3 restores microglial activation and CNS inflammation after the induction of EAE in Peli1-deficient mice. These findings establish Peli1 as a microglia-specific mediator of autoimmune neuroinflammation and suggest a previously unknown signaling mechanism of Peli1 function.
Figures
Comment in
-
Peli1 sets the CNS on fire.Nat Med. 2013 May;19(5):536-8. doi: 10.1038/nm.3176. Nat Med. 2013. PMID: 23652102 No abstract available.
References
-
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. - PubMed
-
- Miller SD, Karpus WJ, Davidson TS. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 2010;11 Chapter 15, Unit 15. - PubMed
-
- Heppner FL, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005;11:146–152. - PubMed
-
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 2005;81:374–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
