

Resistance to aerobic exercise training causes metabolic dysfunction and reveals novel exercise-regulated signaling networks
- PMID: 23610057
- PMCID: PMC3717870
- DOI: 10.2337/db13-0062
Resistance to aerobic exercise training causes metabolic dysfunction and reveals novel exercise-regulated signaling networks
Abstract
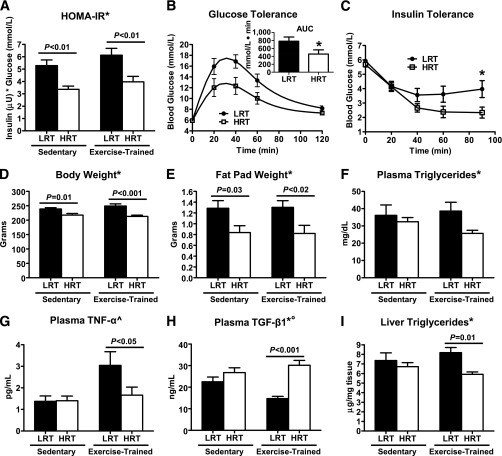
Low aerobic exercise capacity is a risk factor for diabetes and a strong predictor of mortality, yet some individuals are "exercise-resistant" and unable to improve exercise capacity through exercise training. To test the hypothesis that resistance to aerobic exercise training underlies metabolic disease risk, we used selective breeding for 15 generations to develop rat models of low and high aerobic response to training. Before exercise training, rats selected as low and high responders had similar exercise capacities. However, after 8 weeks of treadmill training, low responders failed to improve their exercise capacity, whereas high responders improved by 54%. Remarkably, low responders to aerobic training exhibited pronounced metabolic dysfunction characterized by insulin resistance and increased adiposity, demonstrating that the exercise-resistant phenotype segregates with disease risk. Low responders had impaired exercise-induced angiogenesis in muscle; however, mitochondrial capacity was intact and increased normally with exercise training, demonstrating that mitochondria are not limiting for aerobic adaptation or responsible for metabolic dysfunction in low responders. Low responders had increased stress/inflammatory signaling and altered transforming growth factor-β signaling, characterized by hyperphosphorylation of a novel exercise-regulated phosphorylation site on SMAD2. Using this powerful biological model system, we have discovered key pathways for low exercise training response that may represent novel targets for the treatment of metabolic disease.
Figures





References
-
- Hansen D, Dendale P, van Loon LJ, Meeusen R. The impact of training modalities on the clinical benefits of exercise intervention in patients with cardiovascular disease risk or type 2 diabetes mellitus. Sports Med 2010;40:921–940 - PubMed
-
- Sanz C, Gautier JF, Hanaire H. Physical exercise for the prevention and treatment of type 2 diabetes. Diabetes Metab 2010;36:346–351 - PubMed
-
- Blair SN, Kohl HW, 3rd, Paffenbarger RS, Jr, Clark DG, Cooper KH, Gibbons LW. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA 1989;262:2395–2401 - PubMed
-
- Blair SN, Kampert JB, Kohl HW, 3rd, et al. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA 1996;276:205–210 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DK089503/DK/NIDDK NIH HHS/United States
- NIH5P60DK20572-P/FS/DK/NIDDK NIH HHS/United States
- P30 AG031679/AG/NIA NIH HHS/United States
- 5P30 DK 36836/DK/NIDDK NIH HHS/United States
- R01 DK077200/DK/NIDDK NIH HHS/United States
- P30 DK089503/DK/NIDDK NIH HHS/United States
- P30 DK036836/DK/NIDDK NIH HHS/United States
- T32 GM008322/GM/NIGMS NIH HHS/United States
- P60 DK020572/DK/NIDDK NIH HHS/United States
- R01AR042238/AR/NIAMS NIH HHS/United States
- R24 RR017718/RR/NCRR NIH HHS/United States
- DK068626/DK/NIDDK NIH HHS/United States
- R01 DK068626/DK/NIDDK NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- 1P30AG031679/AG/NIA NIH HHS/United States
- R01 AR042238/AR/NIAMS NIH HHS/United States
- R0D012098A/PHS HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
