

Bats are a major natural reservoir for hepaciviruses and pegiviruses
- PMID: 23610427
- PMCID: PMC3657805
- DOI: 10.1073/pnas.1303037110
Bats are a major natural reservoir for hepaciviruses and pegiviruses
Abstract
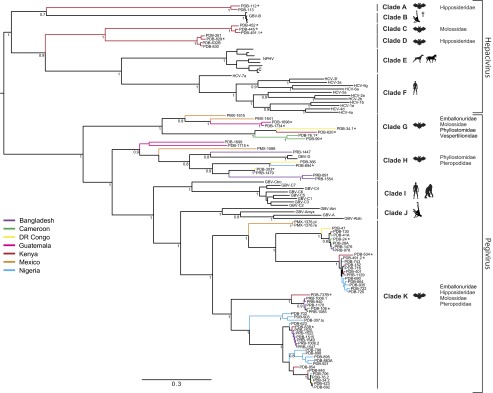
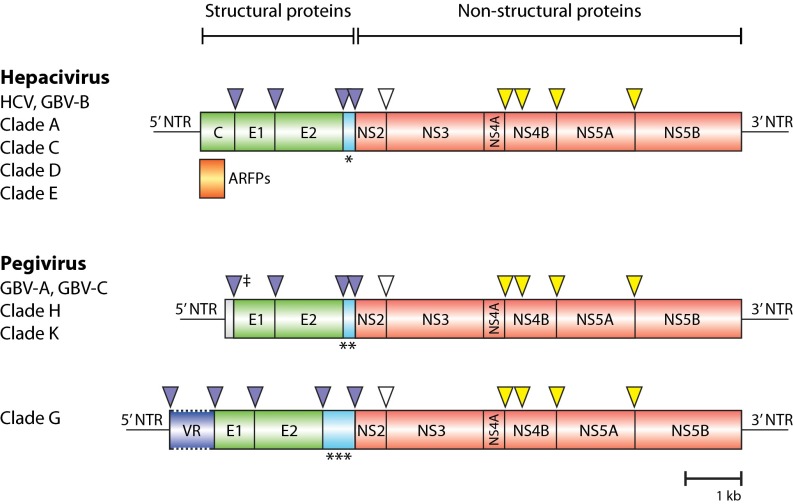
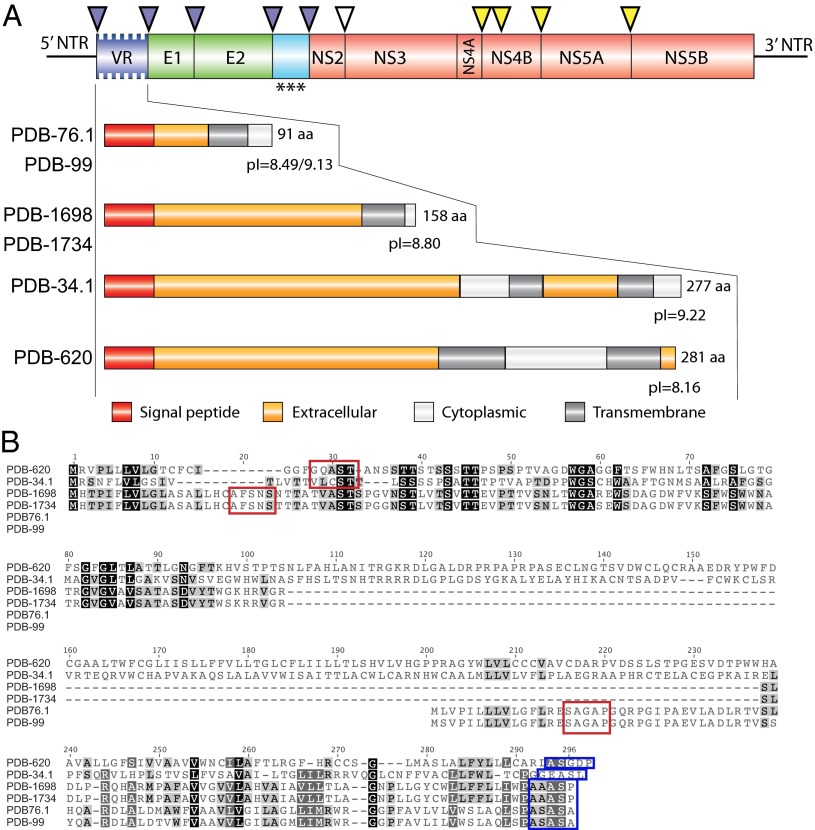
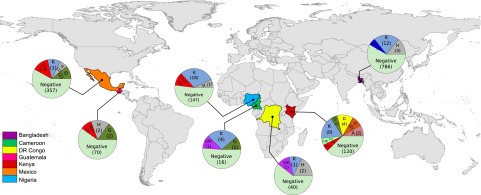
Although there are over 1,150 bat species worldwide, the diversity of viruses harbored by bats has only recently come into focus as a result of expanded wildlife surveillance. Such surveys are of importance in determining the potential for novel viruses to emerge in humans, and for optimal management of bats and their habitats. To enhance our knowledge of the viral diversity present in bats, we initially surveyed 415 sera from African and Central American bats. Unbiased high-throughput sequencing revealed the presence of a highly diverse group of bat-derived viruses related to hepaciviruses and pegiviruses within the family Flaviridae. Subsequent PCR screening of 1,258 bat specimens collected worldwide indicated the presence of these viruses also in North America and Asia. A total of 83 bat-derived viruses were identified, representing an infection rate of nearly 5%. Evolutionary analyses revealed that all known hepaciviruses and pegiviruses, including those previously documented in humans and other primates, fall within the phylogenetic diversity of the bat-derived viruses described here. The prevalence, unprecedented viral biodiversity, phylogenetic divergence, and worldwide distribution of the bat-derived viruses suggest that bats are a major and ancient natural reservoir for both hepaciviruses and pegiviruses and provide insights into the evolutionary history of hepatitis C virus and the human GB viruses.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Teeling EC, et al. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science. 2005;307(5709):580–584. - PubMed
-
- Newman SH, Field HE, de Jong CE, Epstein JH, editors. eds (2011) Investigating the Role of Bats in Emerging Zoonoses: Balancing Ecology, Conservation and Public Health Interests (FAO Animal Production and Health Manual No. 12) (Food and Agriculture Organization of the United Nations, Rome)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
