

Magnetic resonance and computed tomography imaging of the structural and functional changes of pulmonary arterial hypertension
- PMID: 23612440
- PMCID: PMC3903366
- DOI: 10.1097/RTI.0b013e31828d5c48
Magnetic resonance and computed tomography imaging of the structural and functional changes of pulmonary arterial hypertension
Abstract
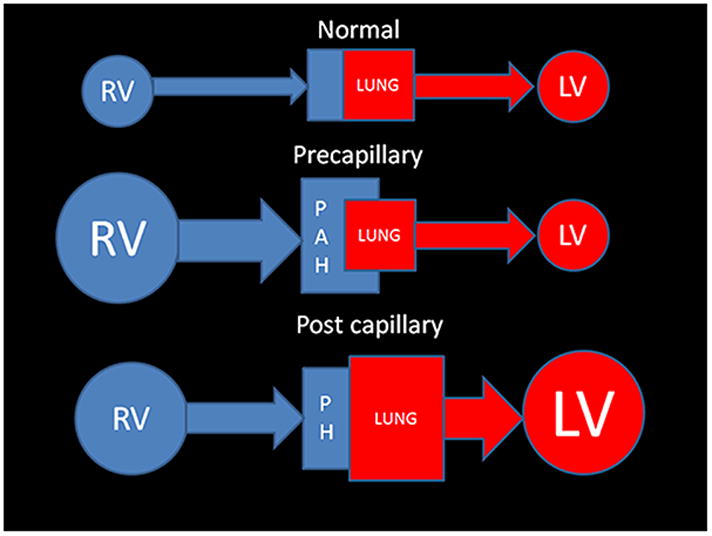
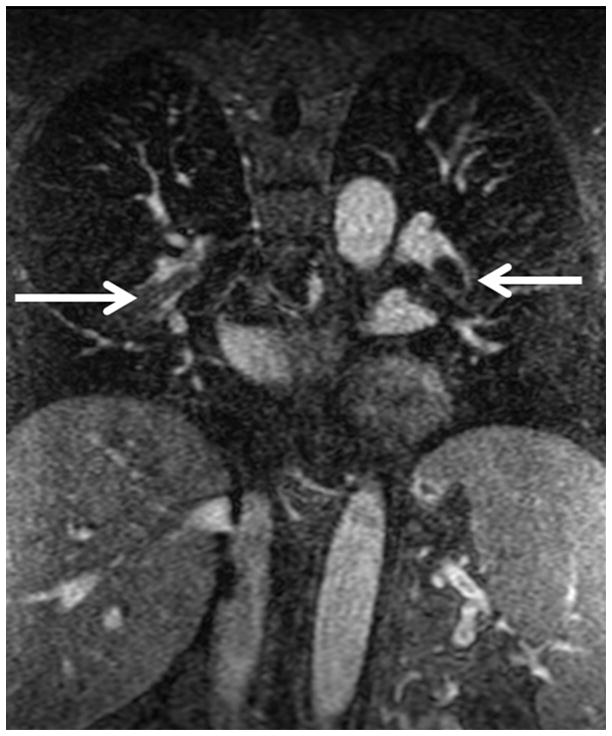
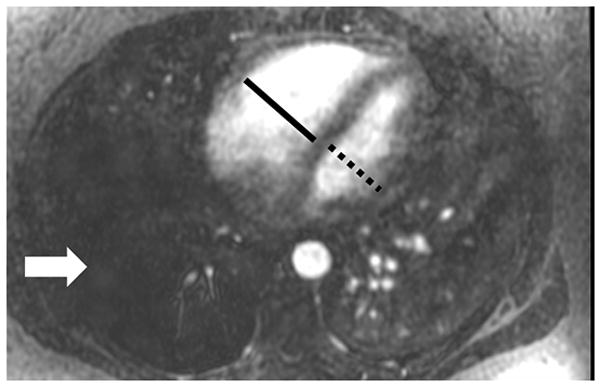
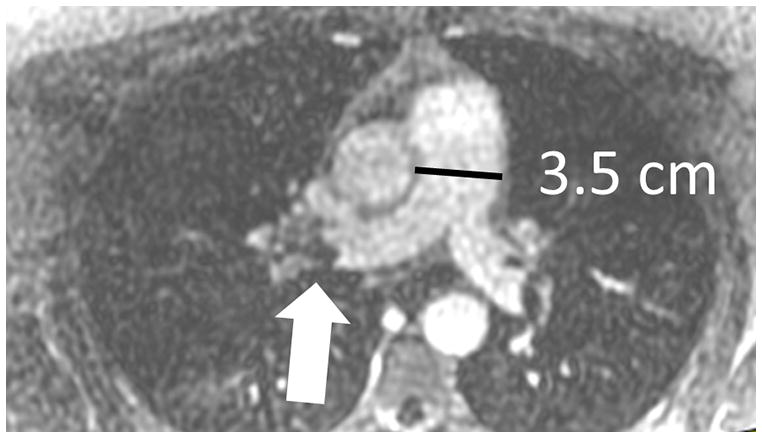
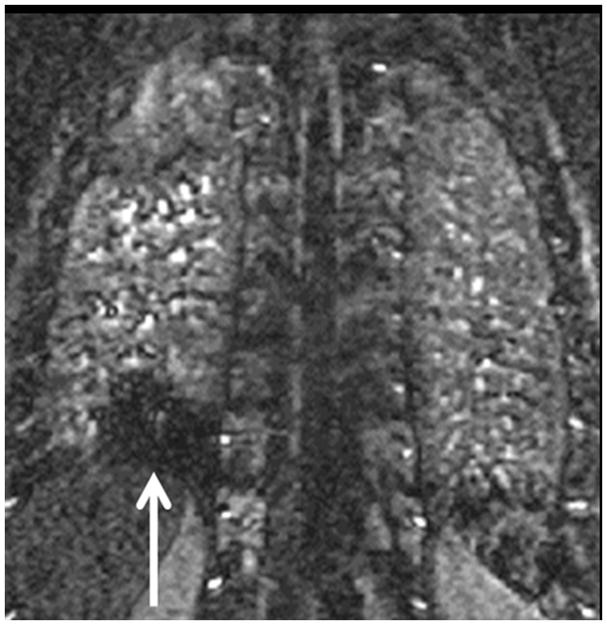
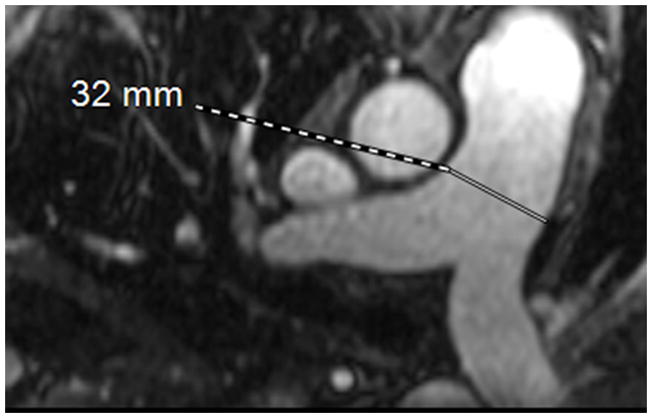
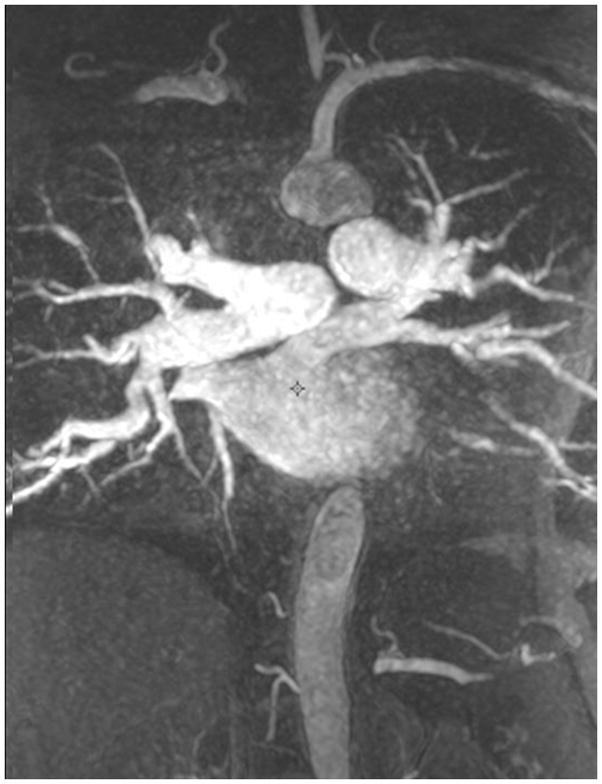
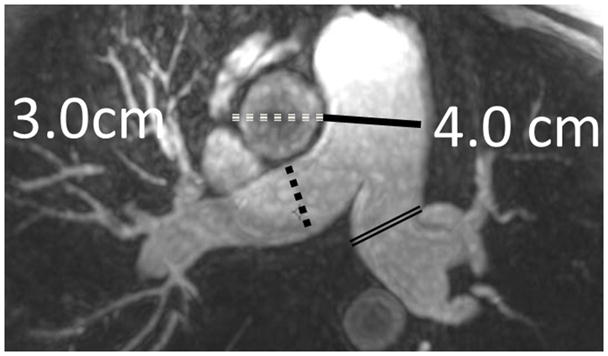
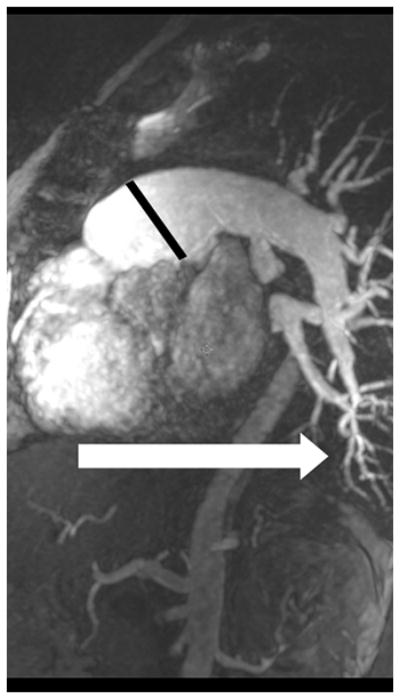
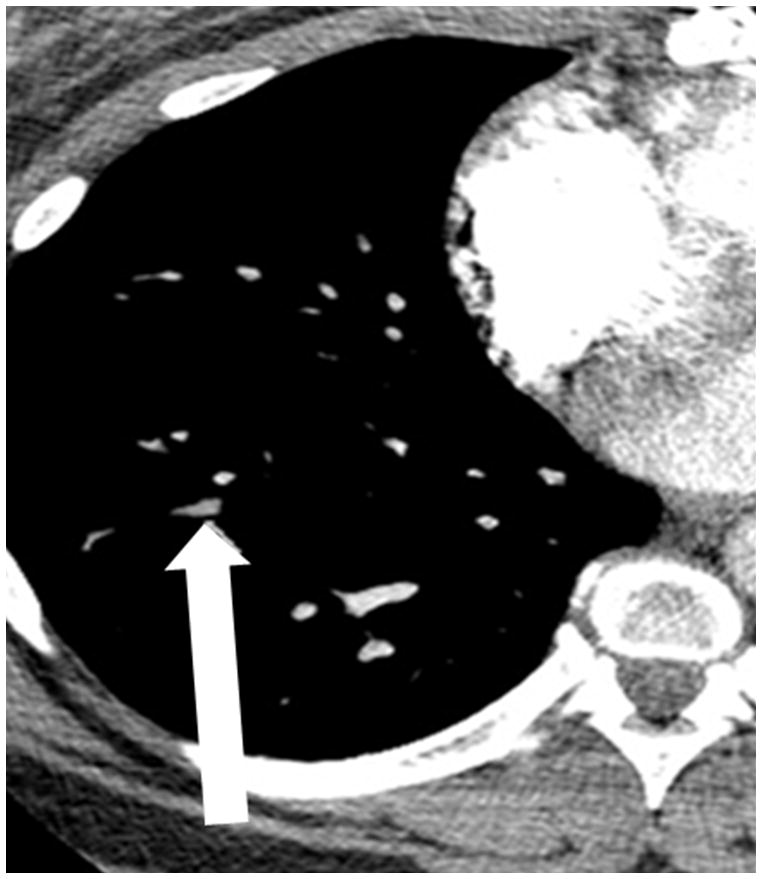
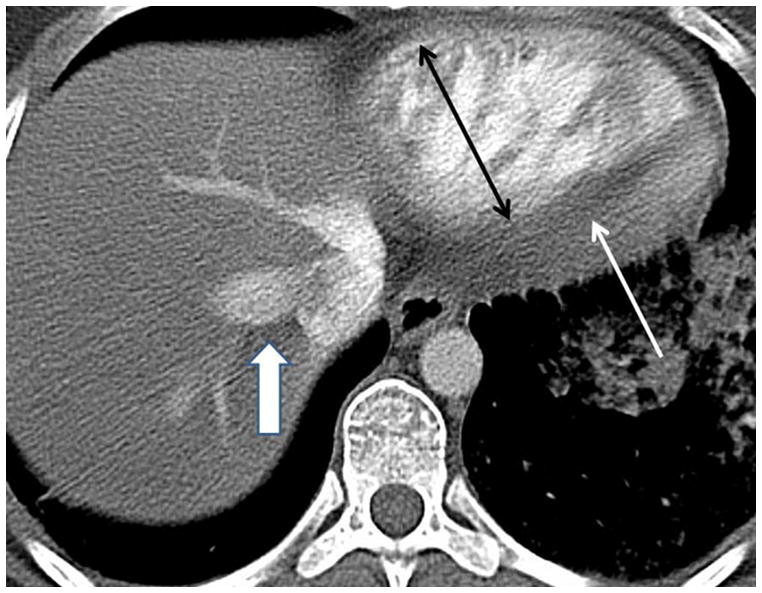
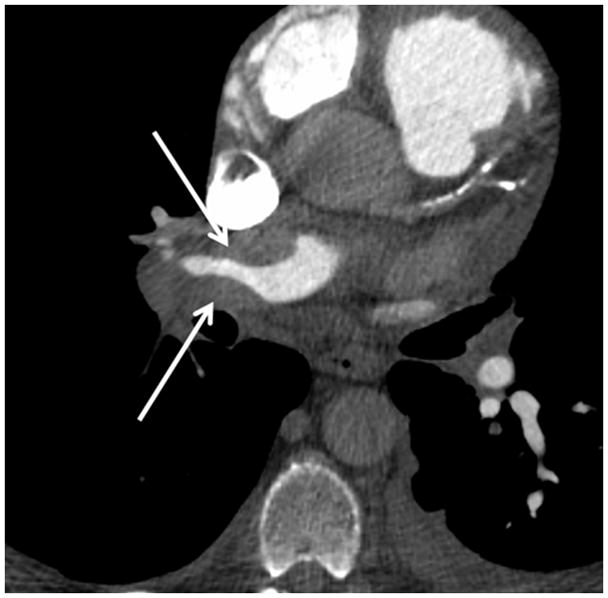
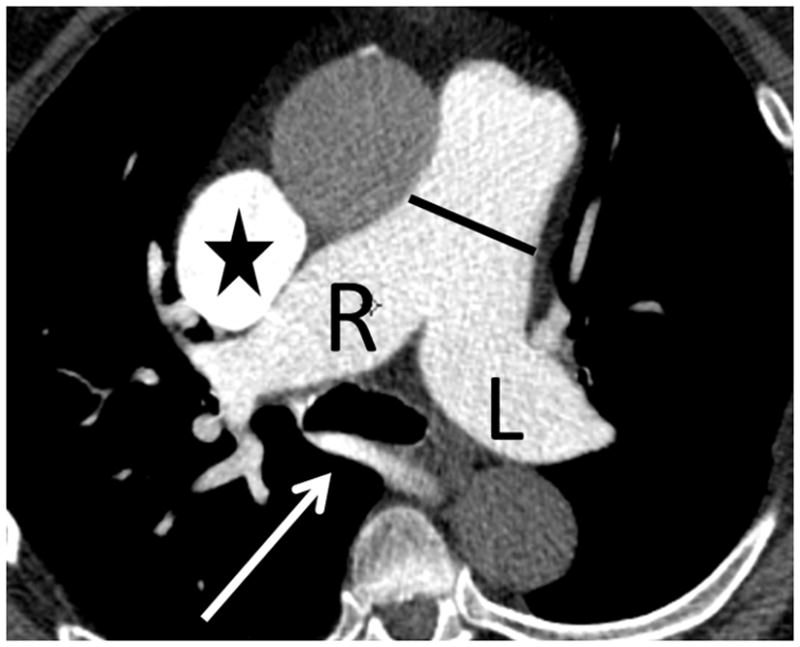
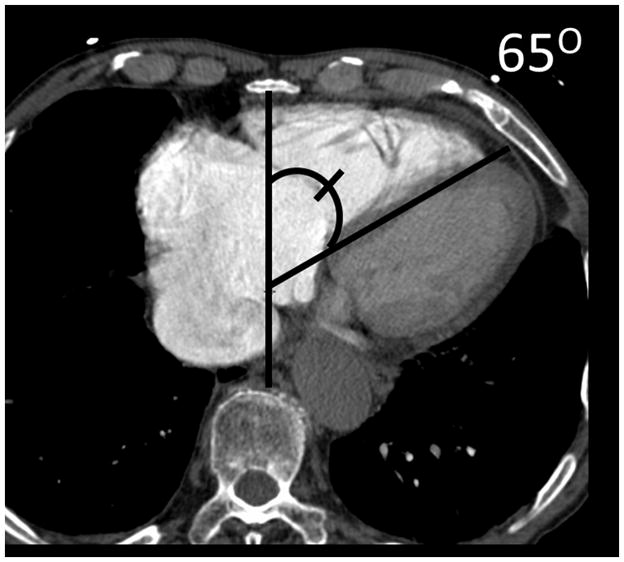
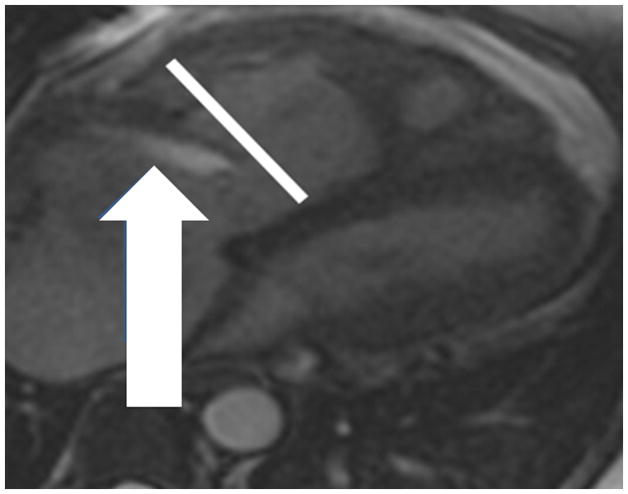
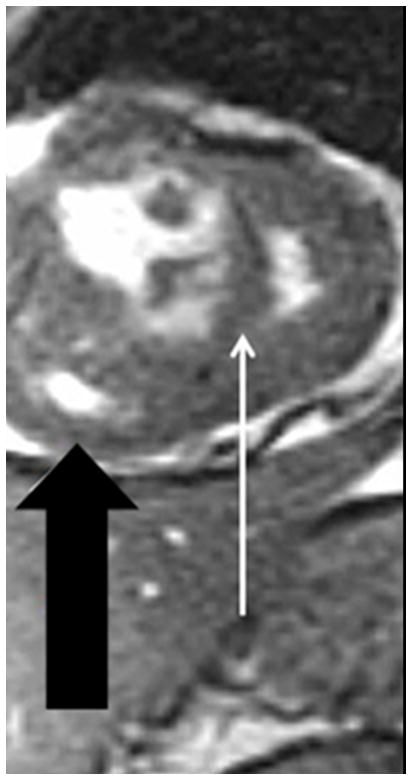
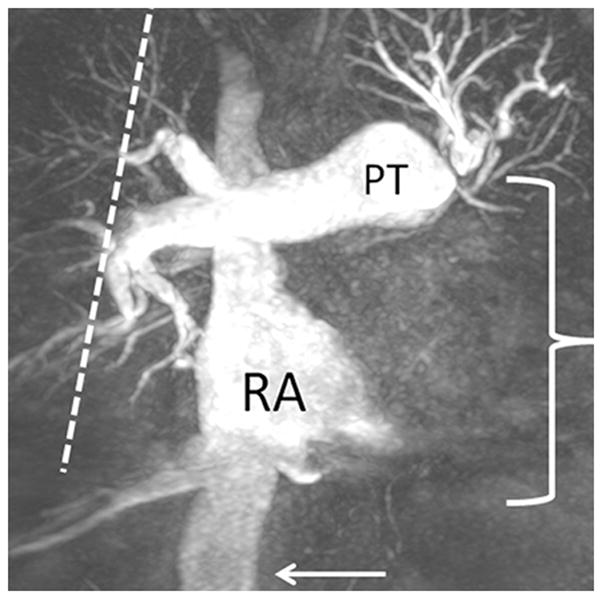
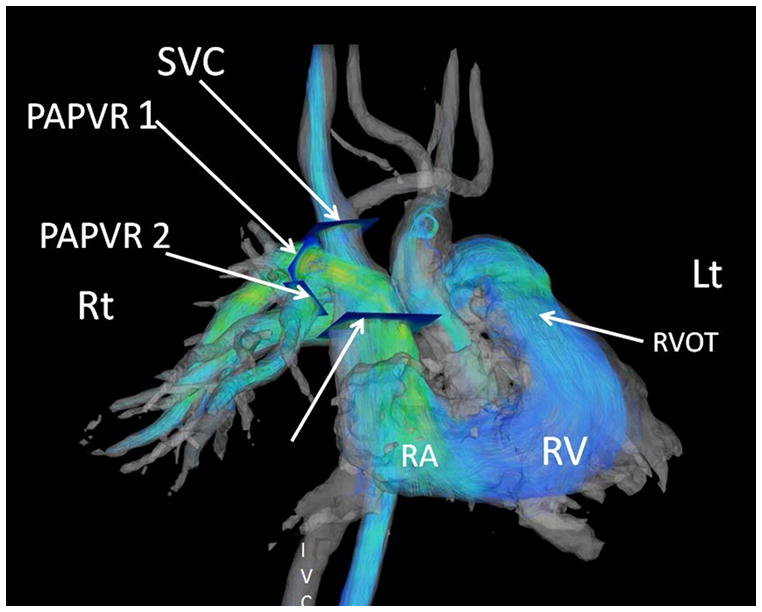
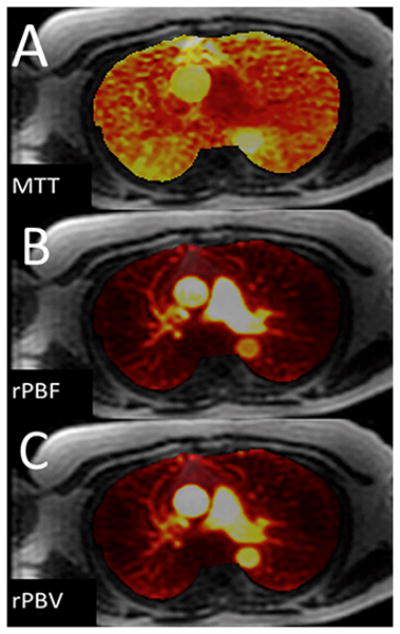
The current Dana Point Classification system (2009) distinguishes elevation of pulmonary arterial pressure into pulmonary arterial hypertension (PAH) and pulmonary hypertension. Fortunately, PAH is not a common disease. However, with the aging of the First World's population, heart failure has become an important outcome of pulmonary hypertension, with up to 9% of the population involved. PAH is usually asymptomatic until late in the disease process. Although features that are indirectly related to PAH are found on noninvasive imaging studies, its diagnosis and management still require right heart catheterization. Imaging features of PAH include the following: (1) enlargement of the pulmonary trunk and main pulmonary arteries; (2) decreased pulmonary arterial compliance; (3) tapering of the peripheral pulmonary arteries; (4) enlargement of the inferior vena cava; and (5) increased mean transit time. The chronic requirement to generate high pulmonary arterial pressure measurably affects the right heart and main pulmonary artery. This change in physiology causes the following structural and functional alterations that have been shown to have prognostic significance: relative area change (RAC) of the pulmonary trunk, right ventricular stroke volume index, right ventricular stroke volume, right ventricular end-diastolic volume index, left ventricular end-diastolic volume index, and baseline right ventricular ejection fraction <35%. All of these variables can be quantified noninvasively and followed up longitudinally in each patient using magnetic resonance imaging to modify the treatment regimen. Untreated PAH frequently results in rapid clinical decline and death within 3 years of diagnosis. Unfortunately, even with treatment, fewer than half of these patients are alive at 4 years.
Figures
References
-
- Humbert M. The burden of pulmonary hypertension. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2007;30(1):1–2. - PubMed
-
- Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2009;54(1 Suppl):S43–54. - PubMed
-
- Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) European heart journal. 2009;30(20):2493–2537. - PubMed
-
- Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coronary artery disease. 2005;16(1):13–18. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
