

Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling
- PMID: 23612963
- PMCID: PMC3675564
- DOI: 10.1074/jbc.M112.435545
Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling
Abstract
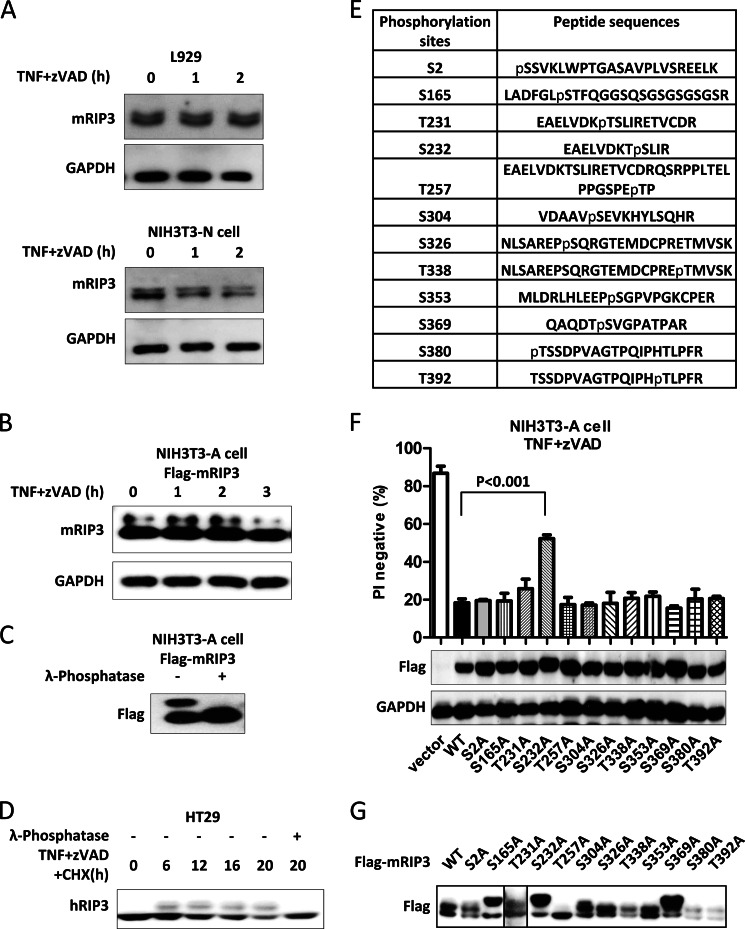
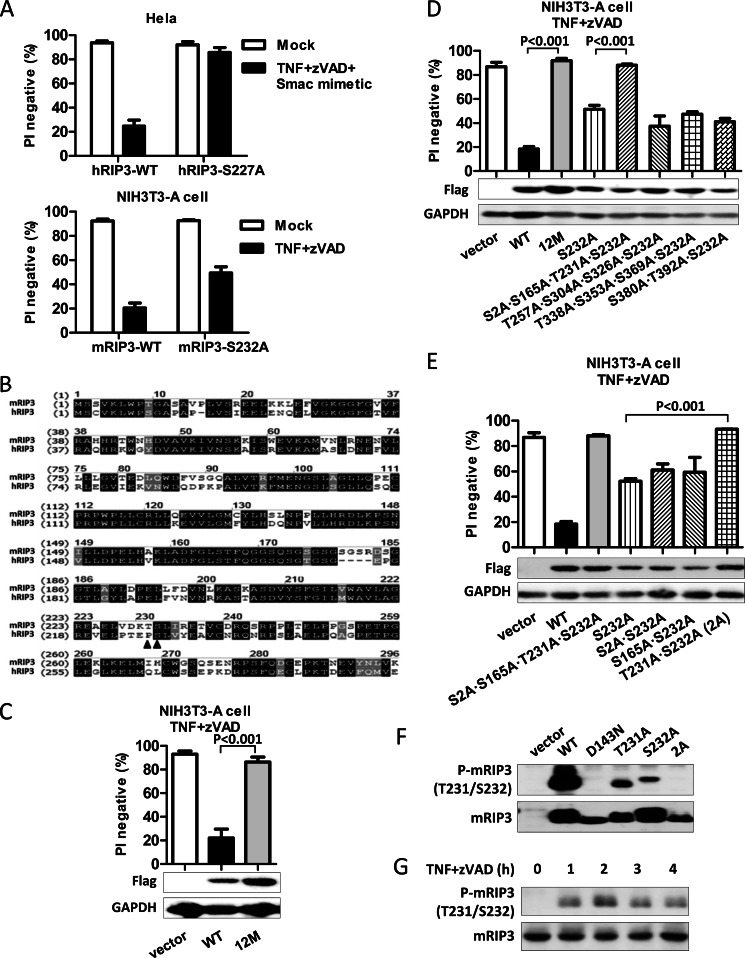
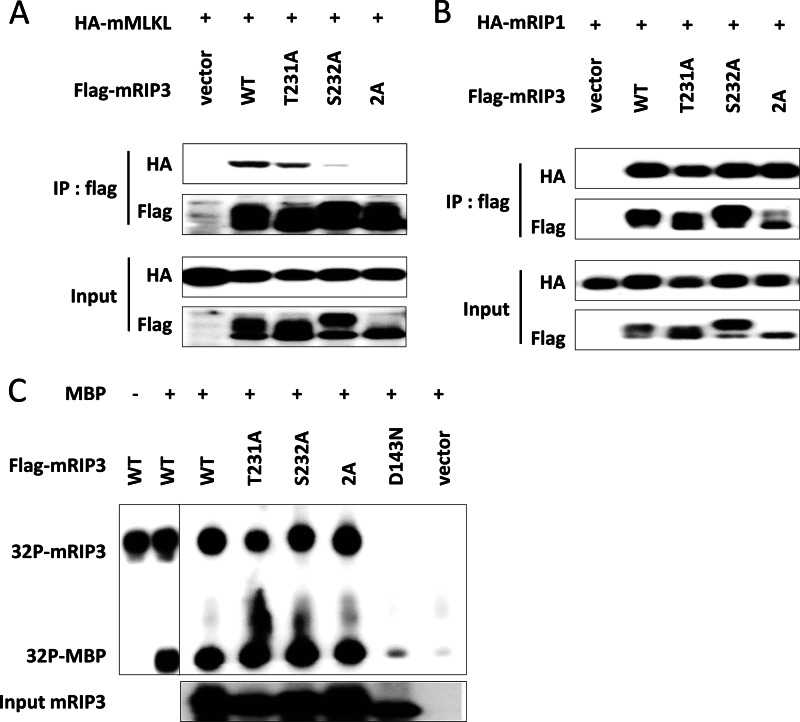
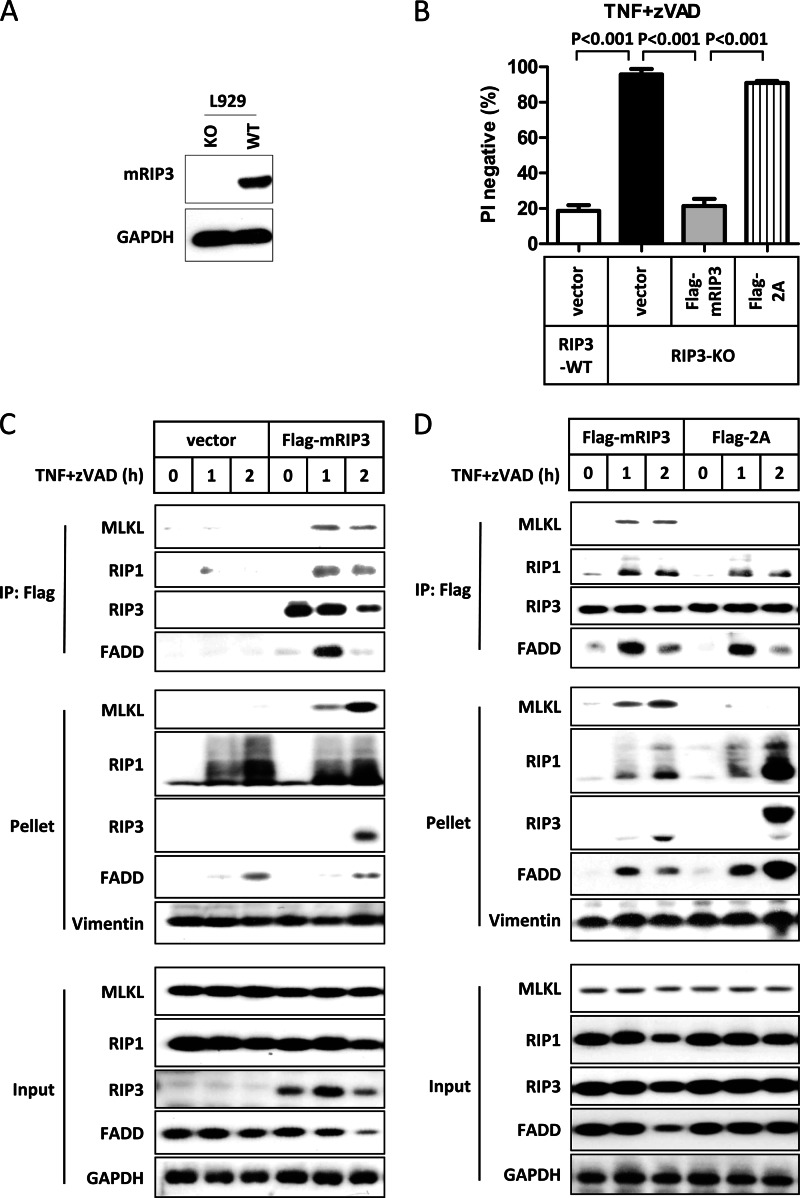
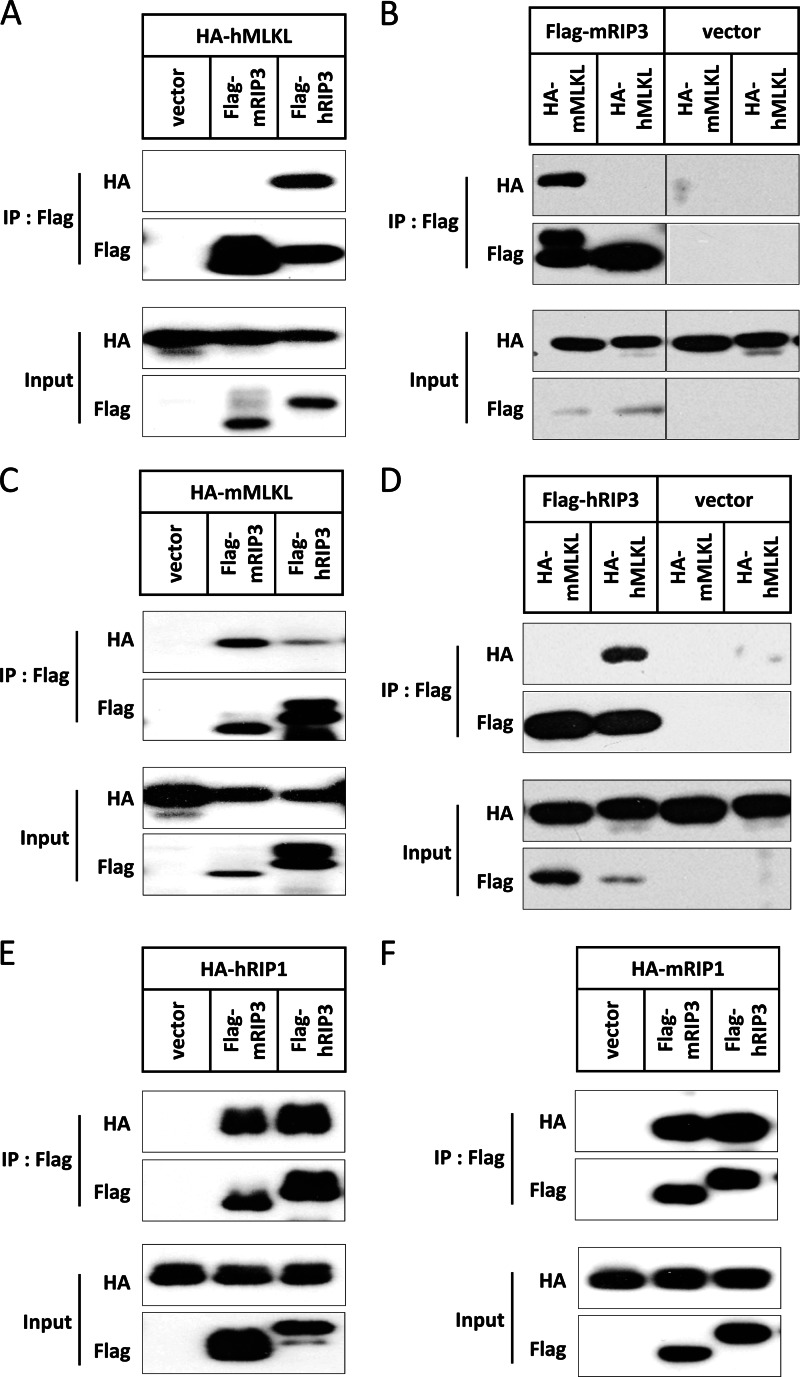
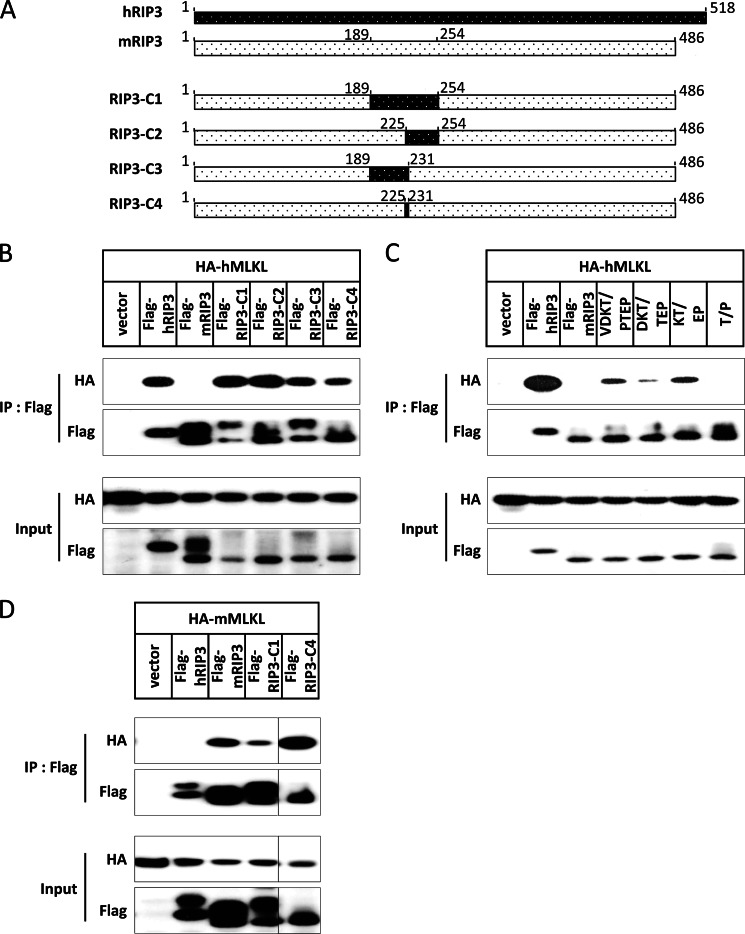
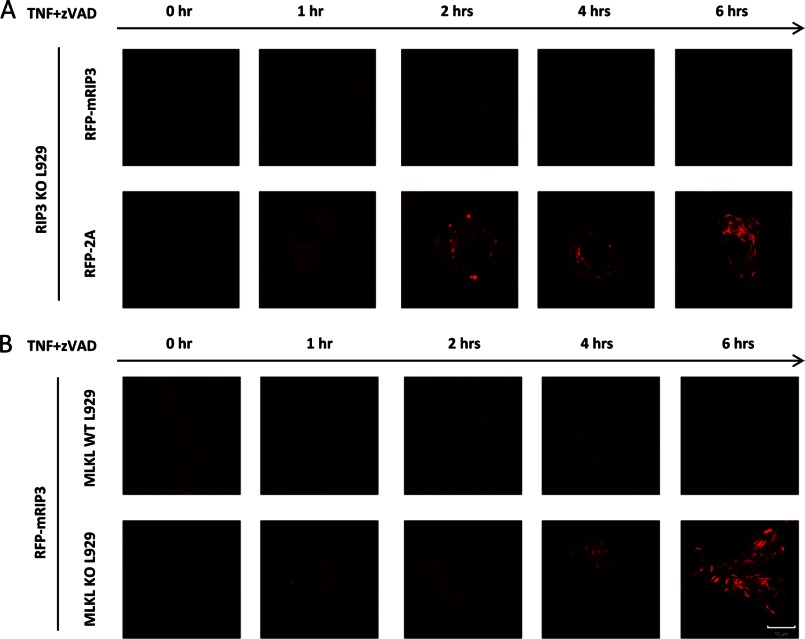
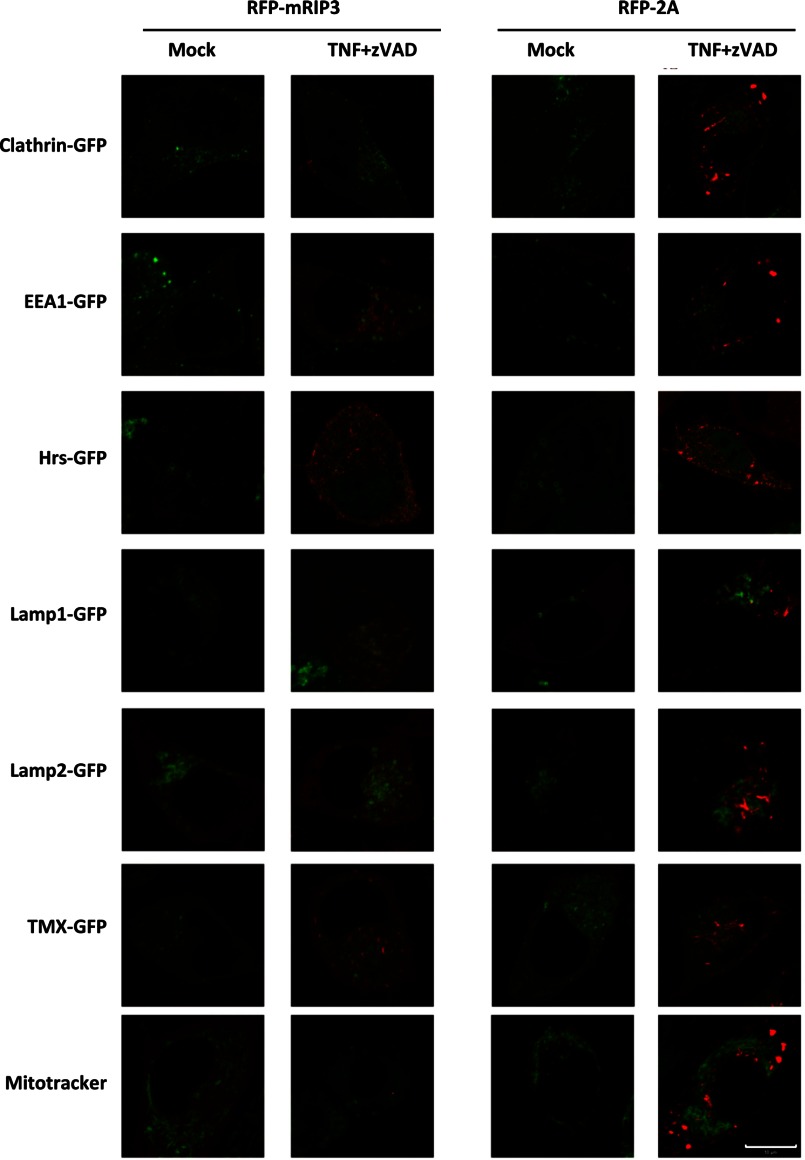
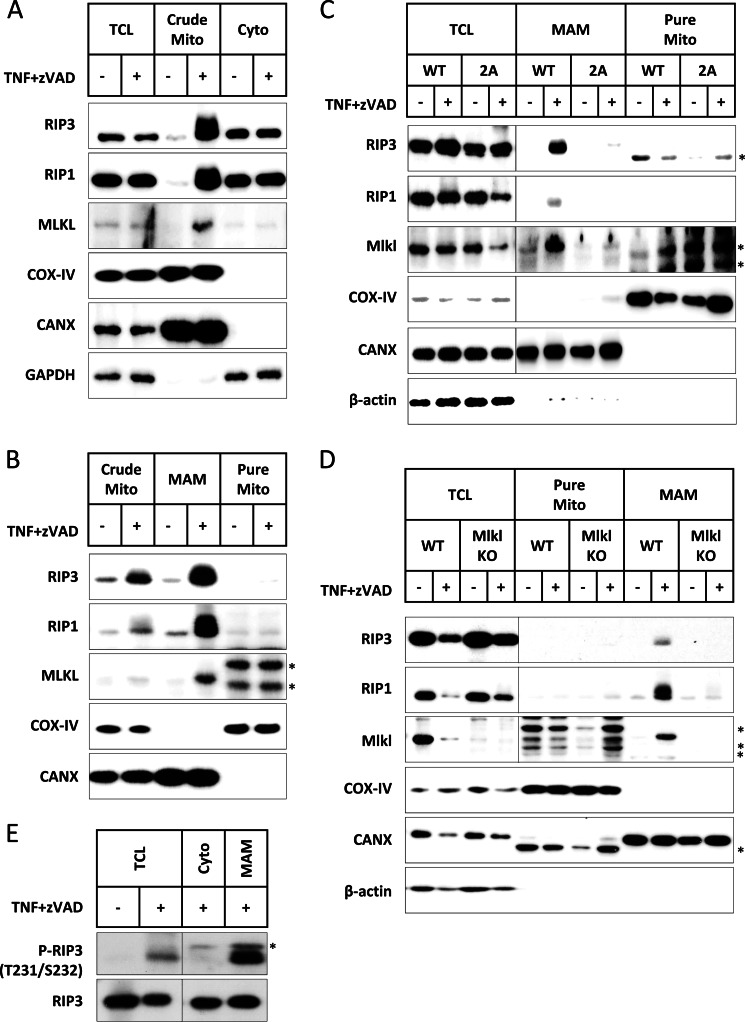
Receptor interacting protein 3 (RIP3) is a protein kinase essential for TNF-induced necroptosis. Phosphorylation on Ser-227 in human RIP3 (hRIP3) is required for its interaction with human mixed lineage kinase domain-like (MLKL) in the necrosome, a signaling complex induced by TNF stimulation. RIP1 and RIP3 mediate necrosome aggregation leading to the formation of amyloid-like signaling complexes. We found that TNF induces Thr-231 and Ser-232 phosphorylation in mouse RIP3 (mRIP3) and this phosphorylation is required for mRIP3 to interact with mMLKL. Ser-232 in mRIP3 corresponds to Ser-227 in hRIP3, whereas Thr-231 is not conserved in hRIP3. Although the RIP3-MLKL interaction is required for necroptosis in both human and mouse cells, hRIP3 does not interact with mMLKL and mRIP3 cannot bind to hMLKL. The species specificity of the RIP3-MLKL interaction is primarily determined by the sequence differences in the phosphorylation sites and the flanking sequence around the phosphorylation sites in hRIP3 and mRIP3. It appears that the RIP3-MLKL interaction has been selected as an evolutionarily conserved mechanism in mediating necroptosis signaling despite that differing structural and mechanistic bases for this interaction emerged simultaneously in different organisms. In addition, we further revealed that the interaction of RIP3 with MLKL prevented massive abnormal RIP3 aggregation, and therefore should be crucial for formation of the amyloid signaling complex of necrosomes. We also found that the interaction between RIP3 and MLKL is required for the translocation of necrosomes to mitochondria-associated membranes. Our data demonstrate the importance of the RIP3-MLKL interaction in the formation of functional necrosomes and suggest that translocation of necrosomes to mitochondria-associated membranes is essential for necroptosis signaling.
Keywords: Cell Death; MLKL; Necroptosis; Necrosis (Necrotic Death); Protein Kinases; Protein Phosphorylation; Protein-Protein Interactions; RIP; RIP3; Tumor Necrosis Factor (TNF).
Figures
Similar articles
-
Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis.Cell Death Differ. 2014 Nov;21(11):1709-20. doi: 10.1038/cdd.2014.77. Epub 2014 Jun 6. Cell Death Differ. 2014. PMID: 24902902 Free PMC article.
-
PI3K mediates tumor necrosis factor induced-necroptosis through initiating RIP1-RIP3-MLKL signaling pathway activation.Cytokine. 2020 May;129:155046. doi: 10.1016/j.cyto.2020.155046. Epub 2020 Feb 28. Cytokine. 2020. PMID: 32114297
-
Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL.J Biol Chem. 2013 Oct 25;288(43):31268-79. doi: 10.1074/jbc.M113.462341. Epub 2013 Sep 9. J Biol Chem. 2013. PMID: 24019532 Free PMC article.
-
More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling.Curr Opin Immunol. 2014 Feb;26:76-89. doi: 10.1016/j.coi.2013.10.017. Epub 2013 Nov 30. Curr Opin Immunol. 2014. PMID: 24556404 Review.
-
Necrosome core machinery: MLKL.Cell Mol Life Sci. 2016 Jun;73(11-12):2153-63. doi: 10.1007/s00018-016-2190-5. Epub 2016 Apr 5. Cell Mol Life Sci. 2016. PMID: 27048809 Free PMC article. Review.
Cited by
-
Regulation of tumour necrosis factor signalling: live or let die.Nat Rev Immunol. 2015 Jun;15(6):362-74. doi: 10.1038/nri3834. Nat Rev Immunol. 2015. PMID: 26008591 Review.
-
An outline of necrosome triggers.Cell Mol Life Sci. 2016 Jun;73(11-12):2137-52. doi: 10.1007/s00018-016-2189-y. Epub 2016 Apr 6. Cell Mol Life Sci. 2016. PMID: 27052312 Free PMC article. Review.
-
ZBP1-Mediated Necroptosis: Mechanisms and Therapeutic Implications.Molecules. 2022 Dec 21;28(1):52. doi: 10.3390/molecules28010052. Molecules. 2022. PMID: 36615244 Free PMC article. Review.
-
Multitasking Kinase RIPK1 Regulates Cell Death and Inflammation.Cold Spring Harb Perspect Biol. 2020 Mar 2;12(3):a036368. doi: 10.1101/cshperspect.a036368. Cold Spring Harb Perspect Biol. 2020. PMID: 31427374 Free PMC article. Review.
-
Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells.Redox Biol. 2018 Oct;19:375-387. doi: 10.1016/j.redox.2018.09.007. Epub 2018 Sep 7. Redox Biol. 2018. PMID: 30237126 Free PMC article.
References
-
- Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
